
改性黄铁矿烧渣催化臭氧氧化水中活性黑5
2015-07-31 07:56何宏平吴德礼马鲁铭吕亚平
同济大学学报(自然科学版) 2015年11期
何宏平,吴德礼,马鲁铭,吕亚平
(1.同济大学 污染控制与资源化研究国家重点实验室,上海200092;2.普莱克斯(中国)投资有限公司,上海201204)
随着染料合成技术的进步,越来越多的新型染料被应用于纺织印染业.以偶氮键(N=N)为特征的偶氮染料在纺织品着色中应用最为广泛[1],但在染色过程中有10%~15%的染料损失会直接进入污水处理系统[2].虽然生物处理法成本低廉,但是对染料中分子复杂的苯环结构的处理效果很不理想,特别是对于深度处理,传统生物方法无能为力[3].
催化臭氧氧化以其强氧化性和较好的可操作性,被广泛应用于对各种难降解有机污染物的处理[4-6].与均相催化臭氧氧化相比,除了能更加高效地分解O3产生HO·外,非均相催化臭氧氧化的反应过程中仅有少量的金属离子溶出,很少或基本不存在金属离子的二次污染,而且性能稳定的非均相催化剂能够重复利用.因此,近年来催化臭氧化主要集中于非均相催化臭氧氧化,其核心之一即寻找高效稳定的固相催化剂,比如活性炭[4]、金属氧化物[5]和负载型催化剂[7]及其改性方法.负载型催化剂中负载金属离子与载体间存在协同作用,Ce是臭氧氧化的有效催化剂,负载Ce后能够显著地提高负载型催化剂的催化活性[6].铁氧化物、锰氧化物等金属氧化物被广泛用于催化臭氧反应,取得了较好的效果[5,7-8].黄铁矿烧渣(PyC)是利用黄铁矿(FeS2)焙烧法生产硫酸时产生的化工废渣,其主要物相组成为铁氧化物,并含有少量的锰、镍、铝、钙、镁等其他金属氧化物,预计对于臭氧具有较好的催化效果,但利用Py C催化臭氧的反应还未见报道.本文以RB5(活性黑5)模拟染料废水为处理对象,研究以负载Ce前后的PyC为催化剂催化臭氧氧化降解有机污染物的可行性.
1 实验部分
1.1 实验试剂与材料
主要试剂:NaOH,浓 H2SO4,Na2S2O7,KI,Ce(NO3)·H2O(均为分析纯);工业级可溶性淀粉,活性黑5(RB5)(国药集团);工业级高纯氧(上海春雨气体)等.主要材料为黄铁矿烧渣(Py C),颗粒比较均匀,200目以下约占70%,黑褐色,来源于江苏某化工厂,其XRD(X射线衍射)图如图1所示.Py C中含Fe质量分数为55.8%,主要以Fe3O4,Fe2O3的形式存在,SiO2的含量也较多,且含有少量的Ca,Mg,Al,Mn等其他金属氧化物.
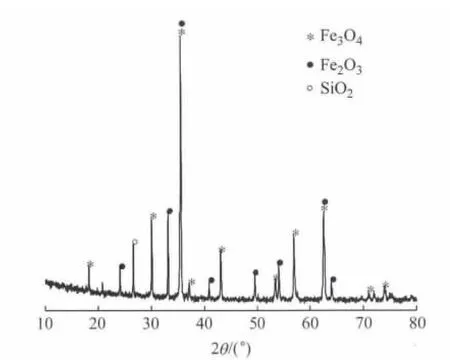
图1 Py C的XRD图Fig.1 XRD pattern of Py C
1.2 改性黄铁矿烧渣制备方法
负载铈改性催化剂的制备采用浸渍法.将过100目筛去除大颗粒的PyC颗粒用超纯水清洗3次;再将洗净后的烧渣(10 g)加入到含有 Ce(NO3)(0.1 mol·L-1,1 L)溶液中慢速搅拌浸渍12 h;此后,沉淀分离,将在80℃下烘干的沉淀物于600℃焙烧6 h,冷却后即得负载铈改性的黄铁矿烧渣(Ce-PyC).
1.3 实验方法和分析方法
实验方法:反应器为圆柱形有机玻璃柱,有效容积为2.2 L,实验前将反应器用去离子水清洗数次,臭氧的产生源气体为工业级高纯氧,臭氧与氧气的混合气体用布于反应器底部的曝气滤头RB5分散.废水采用在实验室自行配制的模拟废水,取2 L(200 mg·L-1)RB5废水加入到反应器中,加入一定量的Ce-PyC或Py C作为催化剂,待臭氧发生器臭氧产生量稳定后通入O3,臭氧的稳定通入量为5 mg·min-1,用机械搅拌使溶液、催化剂和臭氧混合均匀.按不同的时间间隔取样,水样经离心分离可能悬浮的固体后分析污染物浓度、废水TOC以及p H等.
分析方法:Py C的称量用电子天平(PM 400,Mettler),比表面积用比表面积仪测定(JW-BF270,JW),RB5(包括原染料废水,特征波长595 nm)稀释5倍用紫外可见分光光度计法测定(TU-1810,Persee),废水的p H 用p H 计测定(p H-2s,Hach),废水中 TOC用总有机碳分析仪测定(TOC-L CPHCN 200,Shimadzu),PyC的XRD图用X射线衍 射 仪 获 得(D 8 Advance X-ray Spectrometer,Bruker),采用碘量法测O3浓度,溶出的金属离子用ICP方法测定(Agilent 720 ES,Agilent).
2 结果与讨论
2.1 PyC负载Ce工艺条件优化及性能表征
2.1.1 PyC负载Ce工艺条件优化
Ce-PyC催化剂的制备采用初湿含浸法,对浸渍时间、煅烧时间和煅烧温度等关键因素进行了优化,见表1.可以看出,PyC负载Ce最佳的工艺条件为:浸渍时间4 h,煅烧时间6 h,煅烧温度600℃.

表1 Py C负载Ce工艺条件优化Tab.1 Process conditions optimization of Ce-Py C
2.1.2 PyC和Ce-PyC性能的表征
图2,3分别是Py C和Ce-Py C的SEM(扫描电子显微镜)照片和EDS(X射线能谱仪)元素分析图.PyC与Ce-PyC呈现出了不同的表面形态.Py C表面多呈现不规则的片状且稀疏地分布有针状的细小颗粒,而Ce-Py C表面多呈圆形或椭圆形细小颗粒,且分布均匀,可能是因为高温下金属氧化物的形貌改变以及PyC表面沉积了Ce氧化物所致.对PyC和Ce-PyC的EDS元素分析表明,检测到PyC表面Ce元素的存在,证明Ce能有效地负载于PyC的表面.由于PyC在煅烧过程中表面氧化,使得改性后Ce-Py C的氧元素相对含量明显增加.


催化剂表面特性是决定催化剂催化活性的关键影响因素之一,一方面催化剂表面的质子转移会引起水溶液p H的变化,另一方固体催化剂能够直接引发溶解性臭氧的分解[9].用Zeta电位表征PyC负载Ce前后的表面酸碱性(图4),可以看出,PyC与Ce-PyC的零电荷点(p Hpzc)分别为p H = 6.97和7.63,PyC负载Ce改变了催化剂表面的酸碱性,这将会显著地影响黄铁矿烧渣的催化活性.
用 BET(Brunauter-Emmett-Teller)方法分别测试了Py C和Ce-Py C的比表面积ABET(表2).负载Ce降低了PyC的比表面积,这可能是因为煅烧引起PyC表面的侵蚀和坍塌,同时负载于表面的Ce氧化物也会覆盖表面孔道和缝隙从而导致PyC比表面积降低.绝大多数的负载型催化剂经负载后比表面 均 有 所 降 低,如 Mn O/GAC[7],TiO/Al O[10]x223等.此外,测定了PyC负载Ce前后的饱和磁化强度Ms(表2).PyC的饱和磁化强度在负载Ce后有明显的降低,主要是因为有氧条件下的高温煅烧将Fe3O4转化为Fe2O3,但是Ce-PyC的磁性和密度能够足以保证其固液分离.
2.2 负载Ce对PyC催化性能的影响
Ce及其氧化物对臭氧氧化去除污染物具有较好的催化作用[11].因此,为了证实Ce是Ce-PyC催化活性提高的关键因素,还制备了不添加Ce的改性烧渣(m-PyC),其浸渍时间、煅烧时间和煅烧温度均和Ce-PyC制备工艺条件一致,只是浸渍液采用等量超纯水.

图4 Py C和Ce-Py C的Zeta电位Fig.4 Zeta potential of Py C and Ce-Py C

表2 Py C负载Ce前后比表面积及饱和磁化强度Tab.2 BET and Ms of Py C and Ce-Py C
分别研究了PyC,Ce-PyC和m-PyC的催化臭氧氧化效果.如图5所示,3种催化剂都能提高臭氧的氧化效果.O3/Ce-PyC催化剂的效果最好,对TOC的去除率比单独O3体系提高了40.0%,比O3/PyC体系提高了22.6%;但是 O3/m-Py C 的催化效果与O3/Py C相差不大,没有明显提高,说明负载Ce对于Py C催化性能提高起了重要作用,Ce是Ce-PyC较之于PyC催化活性显著提高的关键.

图5 Py C经不同改性处理后对臭氧氧化的催化效果Fig.5 Catalytic effect of Py C after different modifications
2.3 Ce-PyC投加量的影响
Ce-PyC主要通过其中所含的有效成分对臭氧氧化起到催化作用,所以其投加量会影响催化效率.为此研究了p H为7时不同Ce-PyC投加量对处理效果的影响(图6).Ce-Py C投加量从1.0 g·L-1增加至2.5 g·L-1,其TOC去除率从63.15%提高至83.26%,但是当其增加至5.0 g·L-1时,TOC去除率降低至70.43%.此外,经过2 h的反应后O3/Ce-PyC体系1.0 g·L-1投加量TOC去除效果优于5.0 g·L-1投加量.加入的固体催化剂一方面促进O3分解产生HO·,另一方面可能也会消耗产生的HO·.随着催化剂投加量的增加,其体系中消耗HO·的能力也随之增强,从而降低体系矿化有机物的能力.因此确定2.5 g·L-1的Ce-PyC为最佳投加量.

图6 投加量对O3/Ce-Py C体系矿化效果的影响Fig.6 Effect of Ce-Py C dosage on mineralization
2.4 p H的影响
p H是O3分解过程最为重要的影响因素之一.水中的OH-会引发O3的分解,水中p H的升高会提高O3分解的速率,从而促进HO·的产生,同时水溶液p H也会显著地影响催化剂的表面带电特性[12]:

当p H<p Hpzc时催化剂表面趋于质子化,带正电;当p H>p Hpzc时催化剂表面趋于去质子化,带负电.
RB5分子带有4个磺酸基和1个氨基,不同的p H水溶液不仅会影响其平衡状态,也会影响Ce-PyC的表面带点特性,进而影响催化剂与染料分子的接触反应[13]:

因此,开展了催化剂投加量为2.5 g·L-1时不同初始p H染料废水降解效果研究(图7,图7纵坐标中c0为RB5染料初始浓度,c为RB5染料浓度).O3/Ce-PyC在p H 为3.0,4.0,5.5,7.0,10.0这5种条件下均维持着高效稳定的脱色效果;而且p H并未对O3/Ce-Py C处理RB5废水的矿化产生明显的影响,TOC去除率在3~10的p H范围内均维持在80%左右.因此,可以认为Ce-Py C对臭氧氧化在较广的p H范围内均维持着稳定高效的催化活性,其高效地促进O3分解产生HO·,从而加速水溶液中有机物的矿化.相关研究表明,初始p H对目标污染物去除和体系矿化效果均有明显的影响,认为p H的升高有利于污染物的矿化,低p H条件下臭氧分子有选择地与有机物分子反应,而高p H有利于臭氧分解产生无选择性的HO·[14];也有研究发现最优的p H并不是越高越好,Liu等[15]发现6.8为一种Fe-Cu-O催化剂催化臭氧化降解酸性红B的最佳p H.但是基本不受p H影响的非均相催化臭氧氧化体系未见报道,因此,这也是Ce-PyC催化臭氧氧化的最大特色和优势,克服了传统催化臭氧氧化受p H影响大的局限和不足,但其反应机制仍需要进一步深入研究.

图7 p H对Ce-Py C催化臭氧氧化处理效果的影响Fig.7 Effect of p H on catalytic ozonation
2.5 Ce-PyC的重复利用
Ce-PyC的制备方法是将PyC于含有Ce(NO3)的水溶液中浸渍然后高温煅烧,因此Ce,Fe,Mn等金属离子可能会在反应中从催化剂表面溶出,从而降低Ce-PyC的催化活性,缩短催化剂的使用寿命.为了评价催化剂的稳定性,进行了离子溶出测试实验和催化剂的多次重复利用实验.经检测,反应后溶液中常见金属离子如Ce,Fe,Mn的质量浓度均低于仪器的检测线(4μg·L-1),从离子溶出的角度可以认为Ce-PyC性能较稳定.
实验中Ce-PyC重复利用了6次(图8),每次利用后的Ce-PyC经80℃烘干后备用.重复利用3次后Ce-PyC催化活性有稍许的降低,具体表现为TOC去除率从第1次的83.26%降低至第3次的75.10%,但是从第3次开始体系的TOC去除率一直在75%左右,基本保持不变.Ce-PyC催化性能的降低一部分可能是因为重复利用后催化剂表面性能的改变,也可能是因为催化剂表面金属离子的部分损失,尤其是负载的Ce离子,其在PyC表面的负载量很少,溶出的量也很少,但是其对Ce-Py C的催化活性是至关重要的.

图8 Ce-Py C重复利用的催化性能Fig.8 STability of Ce-Py C catalytic activity
3 O3/Ce-PyC去除RB5机理及反应动力学
3.1 HO·产生机理探讨
虽然有研究提出非均相催化臭氧氧化的其他机理,如酸性或偏酸性条件下小分子有机酸与金属氧化物表面络合,络合态的有机物因易于被臭氧分子直接氧化而去除[6].利用非极性表面对弱极性有机物和臭氧分子同时富集的特性,提高表面直接氧化的速度.但是绝大多数的非均相催化臭氧氧化遵循HO·产生机理[14-16].本文PyC负载Ce后能够提高催化臭氧矿化RB5的效率,其关键就在于强化了O3分解产生HO·.染料降解的中间产物如乙酸很难被臭氧直接氧化[17](在p H >5时,反应速率常数k<0.04 mol·L-1·s-1),但是 HO·与乙酸的反应速率常数高达106mol·L-1·s-1[18].
由于表面带点电荷并未平衡,水中的金属类氧化物会强烈吸附水分子[19],吸附的水分子会发生如下电离:
≡H2O↔ HO-+ H+
电离后会在催化剂表面形成表面羟基,表面羟基在水/氧化物表面与溶解O3分子作用生成的自由基能够在氧化物表面和水溶液中引起一系列的链式反应,最终导致 HO·的生成[11].Zhang等[20]已经通过研究证实了表面羟基在催化臭氧氧化去除硝基苯过程中的重要性.马军等[21]也进行了羟基氧化铁催化臭氧分解的机理研究,他们认为羟基氧化铁可以催化臭氧分解产生羟基自由基,氧化物的表面羟基自由基是直接接触水中O3分子的表面物种.
有关水合氧化铁催化O3分子分解产生HO·的机理如下式所示[8,22]:

从2.2得知,Py C对臭氧氧化去除RB5也具有较为显著的催化效果,因此可以借鉴上述HO·的生成机理.
国内外已有关于Ce增强固体催化剂催化性能的研究.Carl等[11]开展了Ce以及Ce与其他金属氧化物复合催化剂对臭氧氧化催化性能的研究,他们认为Ce3+是催化剂催化性能的关键,Ce3+能有效地促进HO·的生成,催化剂含有的Ce3+量越多,催化剂催化性能越高.Alexandra等[23]研究并分析了负载有Ce的活性炭对臭氧氧化处理乙酸和苯胺的催化机理,他们同样认为Ce3+负载量是影响催化剂催化性能的最主要因素,但同时催化剂比表面积和金属氧化物颗粒直径也对负载Ce的活性炭催化性能产生一定的影响.
产生的HO·将会和水中的有机物发生如下的链式反应[24]:

3.2 催化臭氧氧化去除RB5反应动力学机理
O3/Ce-PyC体系去除RB5的机理包括O3分子的直接氧化和O3分解产生HO·的间接氧化,因此RB5染料溶液的脱色可以通过下式表示[4]:
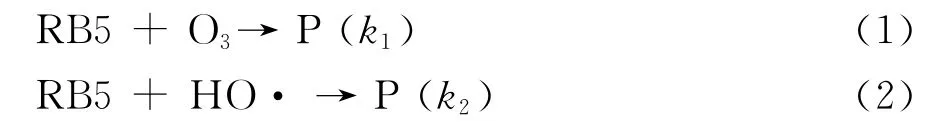
RB5染料的去除情况可以通过下列准一级反应动力学方程表示:

式中:co3为溶解性O3的浓度;cHO·为生成的HO·浓度;k1为RB5分子和O3分子的反应速率常数;k2为RB5分子和HO·的反应速率常数.此时,等式(3)可以通过下列等式表示:


式中:T0为RB5染料初始 TOC大小;k′=k′1cO3+k′2cHO·.
从表3可以看出,O3/PyC,O3/Ce-PyC催化臭氧体系对RB5去除速率常数分别是O3单独氧化的1.40和1.65倍;但 O3/PyC,O3/Ce-PyC催化体系对TOC去除速率常数分别是O3单独氧化的4.44和7.22倍,表明PyC和Ce-PyC对提高有机污染物矿化率具有重要的作用.特别是Ce-Py C展现了更高的催化活性,能明显提高臭氧氧化处理废水过程中TOC的去除,克服了传统的单独臭氧氧化对污染物矿化率低的问题,对于废水深度处理具有优势,能拓展臭氧氧化的应用.
式中:c0为RB5染料初始浓度,k=k1cO3+k2cHO·.类似地,TOC去除情况也可以通过下式表示:
4 结论
采用初湿含浸法制备了负载Ce的PyC作为非均相臭氧氧化的催化剂,通过工艺优化,确定了催化剂制备的最优条件.所制备的Ce-PyC具有优良的催化臭氧效果,特别是对于有机物的矿化率,对TOC的去除速率比单独臭氧提高7.22倍,比未改性的PyC提高4.44倍.而且在p H为3.0~10.0的范围内Ce-PyC均能保持稳定高效的催化活性,TOC去除率均维持在80%左右.解决了臭氧氧化受p H影响大的问题,是该催化剂的最大特色.
催化剂性质稳定,重复利用6次后Ce-PyC的催化活性没有明显的降低,可以重复使用.机理分析表明PyC表面的羟基官能团和负载的Ce是Ce-PyC催化活性提高的关键.
PyC是一种化工废渣,价廉易得,未经处理的PyC对臭氧氧化具有较好的催化性能,而且经简单改性形成的Ce-PyC,其催化性能大大提高.以PyC为催化剂的催化臭氧化技术具有明显的技术经济优势,通过进一步反应条件优化和机理研究有望开发成具有应用前景的新型催化臭氧氧化技术.
[1] Wu J,Wang T.Ozonation of aqueous azo dye in a semi-batch reactor[J].Water Research,2001,35(4):1093.
[2] Gao M,Zeng Z,Sun B,et al.Ozonation of azo dye acid red 14 in a microporous tube-in-tube microchannel reactor:decolorization and mechanism [J].Chemosphere,2012,89(2):190.
[3] Moussavi G,Mahmoudi M.Degradation and biodegradability improvement of the reactive red 198 azo dye using catalytic ozonation with Mg O nanocrystals[J].Chemical Engineering Journal,2009,152(1):1.
[4] Guzman-Perez C A,Soltan J,Robertson J.Kinetics of catalytic ozonation of atrazine in the presence of activated carbon [J].Separation and Purification Technology,2011,79(1):8.
[5] Beltrán F J,Rivas F J.Iron type catalysts for the ozonation of oxalic acid in water[J].Water Research,2005,39(15):3553.
[6] Zhang T,Li W W,CrouéJ.Catalytic ozonation of oxalate with a cerium supported palladium oxide:an efficient degradation not relying on hydroxyl radical oxidation [J].Environmental Science and Technology,2011,45(21):9339.
[7] Sui M H,Liu J,Sheng L.Mesoporous material supported manganese oxides(Mn Ox/MCM-41)catalytic ozonation of nitrobenzene in water [J]. Applied Catalysis B:Environmental,2011,106(1):195.
[8] Zhao L,Ma J,Sun Z Z.Mechanism of heterogeneous catalytic ozonation of nitrobenzene in aqueous solution with modified ceramic honeycomb[J].Applied Catalysis B:Environmental,2009,89(3-4):326.
[9] Stumm W,Morgan J J.Aquatic chemistry:chemical equilibria and rates in natural waters[M].3rd ed.New York:John Wiley and Sons,1981.
[10] Beltran F R,Monterodeespinosa F.A TiO2/Al2O3catalyst to improve the ozonation of oxalic acid in water[J].Applied Catalysis B:Environmental,2004,47(2):101.
[11] Carla A O,JoséJ M,Manuel F R,et al.Ceria and ceriumbased mixed oxides as ozonation catalyst [J]. Chemical Engineering Journal,2012,200-202:499.
[12] Stumm W.Chemistry of the solid-water interface:processes at the mineral-water and particle-water interface in natural systems[M].New York:John Wiley and Sons,1992.
[13] Avramescu S M,Mihalache N,Bradu C,et al.Catalytic ozonation of acid red 88 from aqueous solutions[J].Catalysis Letters,2009,129(3-4):273.
[14] Martins R C,Quinta-Ferreira R M.Catalytic ozonation of phenolic acids over a Mn-Ce-O catalyst[J].Applied Catalysis B:Environmental 2009,90(1-2):268.
[15] Liu X Y,Zhou Z M,Jing G H,et al.Catalytic ozonation of acid red B in aqueous solution over a Fe-Cu-O catalyst [J].Separation and Purification Technology,2013,115(2):129.
[16] Erol F,Özbelge T A.Catalytic ozonation with non-polar bonded alumina phases for treatment of aqueous dye solutions in a semi-batch reactor[J].Chemical Engineering Journal,2008,139(2):272.
[17] HoignéJ,Bader H.Rate constants of reactions of ozone with organic and inorganic compounds in water-Ⅱ:dissociating organic compounds[J].Water Research,1983,17(2):185.
[18] Getoff N,Schwörer F,Markovic V M.Hydroxyl radical induced decomposition of aliphatic acids in oxygenated and deoxygenated aqueous solutions [J].Journal of Physical Chemistry,1971,75(3):749.
[19] Joseph Y,Ranke W,Weiss W.Water on FeO(111)and Fe3O4(111):adsorption behavior on different surface terminations[J].Journal of Physical Chemistry B,2000,104(14):3224.
[20] Zhang T,Ma J.Catalytic ozonation of trace nitrobenzene in water with synthetic goethite [J].Journal of Molecular Catalysis A:Chemical,2008,279(1):82.
[21] 马军,张涛,陈忠林,等.水中羟基氧化铁催化臭氧分解和氧化痕量硝基苯的机理探讨 [J].环境科学,2005,26(2):78.MA Jun,ZHANG Tao,CHEN Zhonglin,et al.The mechanism of ozone decomposition and trace nitrobenzene ozonation catalyzed by ferric hydroxide [J].Environmental Science- China,2005,26(2):78.
[22] Gunten U V.Ozonation of drinking water:partⅠ.oxidation kinetics and product formation[J].Water Research,2003,37(7):1443.
[23] Alexandra G,JoaquíS,Enrique V R,et al.Highly dispersed ceria on activated carbon for the catalyzed ozonation of organic pollutants[J].Applied Catalysis B:Environmental,2012,113-114:308.
[24] Anaid C Q,Carlos B D,Gabriela R,et al. Wastewater ozonation catalyzed by iron [J].Industrial & Engineering Chemical Research,2011,50(5):2488.
猜你喜欢
工业安全与环保(2022年10期)2022-10-28
陶瓷学报(2020年6期)2021-01-26
装备维修技术(2020年5期)2020-11-20
矿产综合利用(2020年1期)2020-07-24
浙江大学学报(理学版)(2020年1期)2020-03-12
中学生数理化·中考版(2018年11期)2019-01-31
教学考试(高考化学)(2018年5期)2018-12-06
分析化学(2018年1期)2018-01-18
分析化学(2017年9期)2017-10-16
天津城建大学学报(2015年5期)2015-12-09
