
脊髓性肌萎缩症2个核心家系SMN1基因分析
2015-10-22 12:44曾光群杨季云张丁丁通讯作者
中国实用神经疾病杂志 2015年23期
曾光群 杨季云 张丁丁(通讯作者) 陈 蓉 李 宁
1)四川彭州市人民医院 彭州 611930 2)四川省医学科学院 四川省人民医院人类疾病基因研究四川省重点实验室 成都 610072 3)川北医学院 南充 637000 4)遵义医学院 遵义 563000
脊髓肌肉萎缩症(spinal muscular atrophy,SMA)是仅次于囊性纤维化的儿童致死性疾病,是由于脊髓前角运动神经元变性引起的渐进性近端肌肉无力和瘫痪的神经肌肉性疾病,大多数为常染色体隐性遗传,也有报道为常染色体显性遗传和X 连锁遗传[1]。发病率1/6 000~1/10 000,携带率在不同的人群中为1/40~1/60[2-3],我国南部正常人群此病的携带率约1/60[4],上海、台湾正常人群此病的携带率约1/39[5]和1/48[6]。
1995年法国学者将SMA 的基因定位于5q11.2~13.3,认为反向重复的运动神经元生存基因(survival motor neuron gene,SMN)为致病基因[7]。该基因有2个序列高度相似的拷贝:靠近端粒的决定性基因SMN1和靠近着丝粒的修饰基因SMN2,两者间有5个碱基的区别,编码序列内仅1 个碱基差异。SMN 所在的染色体区域内结构复杂,且存在众多重复序列和假基因簇,致其结构不稳定,发生缺失或转换的频率较高,使相应的SMN1基因拷贝数复杂多变。95%以上的SMA 患者有SMN1第7号外显子纯合缺失,其余5%是SMN1点突变或者复合型的杂合缺失[1]。SMN2 基因的拷贝数是SMN1基因缺失的剂量补偿,与患儿临床表型的严重程度相关[8]。MLPA 是一种检测基因缺失或重复的高效、准确方法[9],STR 连锁能分析风险染色体的来源。本研究联合两种方法对2个SMA 家系成员SMN1基因检测,明确了基因携带情况,给患儿和家庭提供了完整的评估。
1 资料和方法
1.1 一般资料 先证者1,女,11岁,G1P1,足月顺产,围生期无异常。3岁前运动无异常,自3岁开始出现下蹲困难,易摔等现象,下肢比上肢严重。体格检查:身高130cm,四肢肌肉萎缩,肌力、肌张力及膝腱反射减弱,病理征未引出。头颅MRI未见明显异常,肌电图表现为神经源性损害。辅助检查:磷酸肌酸激酶324U/L(参考值26.00~174.00U/L),磷酸肌酸激酶同工酶52U/L(参考值0~25.00U/L)。患儿父母体健,非近亲结婚,家族中无同类病史。
先证者2,男4岁。G1P1,足月顺产,围生期无异常。患儿自幼运动发育落后,2岁左右开始出现运动能力倒退,走路不稳,上下楼梯困难,下蹲后不易站起,呈鸭子步态。体格检查:体型瘦小,双侧腓肠肌略肥大,四肢肌力Ⅳ级、肌张力降低,膝腱反射减弱,病理征阴性,Gower征(+)。辅助检查:磷酸肌酸激酶408U/L,磷酸肌酸激酶同工酶62U/L,肌电图呈典型的失神经性改变。患儿父母体健,非近亲结婚,家族中无同类病史,母亲怀孕18 周。
1.2 研究方法
1.2.1 基因组DNA 提取:签署知情同意书后,取患儿及家人乙二胺四乙酸钠抗凝外周血2 mL,无菌抽取孕妇羊水5 mL。用DNA 抽提试剂盒(北京天根生化科技有限公司)提取基因组DNA,操作步骤严格按照试剂盒说明书进行。NanoDrop2000测定浓度。将DNA 浓度校正至50ng/μL,-20 ℃保存。
1.2.2 MLPA 检测:按荷兰MRC-Holland公司提供的MLPA 试剂盒检测。DNA 样本(50ng)5μL,经变性、探针杂交过夜、连接和PCR 反应等步骤后,取PCR 产物1.0μL,加1.5μL LIZ-500,HD 7μL,混匀,ABI3130xl测序仪进行片段分析[10]。
1.2.3 结果判定:使用GeneMapper软件收集数据,Coffalyser软件进行分析。如无信号则为外显子缺失,表现为相关峰消失,信号成倍增加表示发生重复。携带者相应外显子缺失信号降低35%~55%。
1.2.4 STR 基 因 连 锁 分 析:根 据 文 献[11],选 用4 个 连 锁STR 位点(D5S435、D5S351、D5S610、D5S629)对2个家系分析。引物由上海生工合成,荧光标记引物两翼,ABI3130xl遗传分析仪上机检测,进行单倍型连锁分析。
2 结果
2.1 MLPA 结果 家系1中先证者SMN1基因第7号外显子及8号外显子拷贝数均为零,母亲和外祖母SMN1基因第7号外显子及8号外显子为1个拷贝数,其爷爷SMN1基因第7号外显子及8号外显子为3个拷贝数。父亲SMN1基因第7号外显子及8号外显子为2个拷贝数。家系2先证者SMN1基因第7号外显子及8号外显子拷贝数均为零,母亲和胎儿SMN1基因第7号外显子及8号外显子为1个拷贝数,父亲SMN1基因第7号外显子及8号外显子为2个拷贝数。
2.2 单倍型分析结果 家系1先证者与其父亲有相同的母源致病的SMN1等位基因。家系2 先证者和胎儿均遗传1条父亲的致病的等位基因片段。

家系1
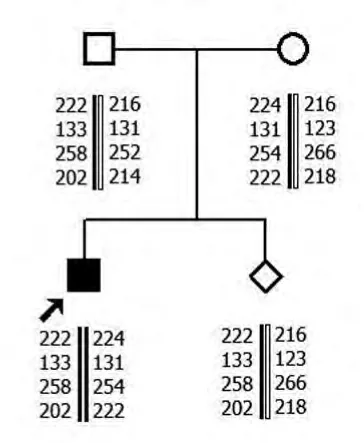
家系2
3 讨论
SMA 的诊断主要依靠临床表现、实验室检查、家族遗传史及基因检测4个方面,但临床表现和常规辅助检测缺乏特异性,故临床上对确诊SMA 更多依赖于基因检测。临床上SMA 各型间的差异很大,依据发病年龄和临床病程严重程度的不同,分为Ⅰ型、Ⅱ型、Ⅲ型和Ⅳ型,其中Ⅰ型约占70%,临 床 表 型 最 严 重,多 数 于6 个 月 内 起 病,2 岁 内 夭 折[12]。SMN1基因缺失(包括SMN1基因纯合缺失或SMN1基因转换为SMN2基因)或SMN1基因内微小突变是引起SMA 的主要原因。SMN1基因缺失及其附近基因的大范围缺失,引起的临床表型较严重,SMN1基因转换为SMN2基因,引起的临床表型较轻,但SMN 基因的缺失和表型的严重程度缺失之间没有相关性[13]。
每条染色体上有0~4个拷贝的SMN 基因,大部分纯合缺失的患者SMN1拷贝数为0,健康人SMN1拷贝数为2,携带者和杂合缺失的患者SMNI拷贝数为1,约5%的正常个体可有3~4个SMN1拷贝,曲晓星等[5]报道在正常个体中有5~7个拷贝。因而通过检测SMN1拷贝数可诊断SMA患者以及筛查携带者。SMA 携带者主要有4种基因型[14]:2条同源染色体上,1 条染色体上有1 拷贝正常的SMN1 基因,另1条缺失SMN1基因的为“1+0”型;1条染色体上有2拷贝SMN1基因,另1条缺失SMNI基因的“2+0”型;1条染色体上有1拷贝SMN1基因,另1条SMN1基因发生微小突变的“1+1m”型;1条染色体上有2拷贝SMNI基因,另1条SMN1基因发生突变的“2+1m”型;“1+0”型携带者最常见,约占95%,其他约占5%。
本研究先证者1和2SMN1基因的第7号外显子及8号外显子拷贝数均为零,可在基因水平诊断先证者为SMA。先证者1母亲和外祖母SMN1基因第7号外显子及8号外显子为1个拷贝数,先证者2母亲SMN1基因第7号外显子及第8号外显子为1个拷贝数,为杂合缺失突变,提示其均为携带者。先证者1爷爷SMN1 基因第7 号外显子及第8号外显子为3个拷贝数,SMN1基因正常。先证者1和2父亲SMN1基因第7号外显子及8号外显子为2个拷贝数,表型上为正常。但其家中却有患儿出现,结合STR 分析,显示先证者遗传了1条父亲致病的等位基因片段,为“2+0”SMA致病基因携带者,即1条染色体上有2拷贝SMN1基因,另1条染色体缺失SMN1基因。家系2胎儿SMN1基因第7号外显子及8号外显子为1个拷贝数,1条为父源致病等位基因片段,1条为母源正常等位基因片段,为SMA 携带者,可以生育。
SMA 的严重程度由两条染色体上携带突变的SMN 基因、SMN2基因拷贝数等其他因素决定。当前检测SMA 携带者的方法,尚存在一定局限性。“1+1m”型和“2+0”型的SMA 携带者每条染色体上含SMN1相同剂量的拷贝数,因此普通人群中检测出含有2拷贝SMN1只能说明携带者的风险降低,不能完全排除“1+1 m”型杂合缺失和“2+0”型SMA 携带者,在下一代中仍可有发病者。由于SMA 是一种发病率较高的遗传性致死性疾病,基因携带率大,至今尚无有效的治疗措施,产前诊断是预防该病的有效手段,因而患者的诊断以及携带者的检出尤为重要。我们联合MLPA 和STR 连锁分析方法对2个核心家系SMN1基因分析,验证了先证者的风险等位基因的来源,检出有2个拷贝的“2+0”型SMA 携带者,增加了检出率。
通过以上分析,我们发现将MLPA 和STR 连锁分析的联合应用,可充分发挥二者的优势,明确SMA 携带者,确诊先证者。临床上考虑为SMA 患者应对以下情况进行SMN检测:(1)有SMA 生育史;(2)自身为SMA 携带者,欲生育,则须对其配偶进行携带者检测;(3)夫妻双方家族均有脊髓性肌肉萎缩症病史;(4)夫妻均为携带者或曾经是脊髓性肌肉萎缩症患者;(5)先证者经基因诊断明确的高危产妇的。为保证产前诊断的准确性,应结合多态性连锁分析以减少风险发生率。对于无SMN1 纯合缺失的患者,对其基因内微小突变检测。对基因确诊的患者,要分析5q13 区域内其他修饰基因的变异,预测临床表型和病情进展。
[1] Jiang W,Ji X,Xu Y,et al.Molecular prenatal diagnosis of autosomal recessive spinal muscular atrophies using quantification polymerase chain reaction[J].Genet Test Mol Biomarkers,2013,17(5):438-442.
[2] Markowitz JA,Singh P,Darras BT.Spinal muscular atrophy:a clinical and research update[J].Pediatr Neurol,2012,46(1):1-12.
[3] Kocheva SA,Plaseska-Karanfilska D,Trivodalieva S,et al.Prenatal diagnosis of spinal muscular atrophy in Macedonian families[J].Genet Test,2008,12(3):391-393.
[4] Prior TW.Spinal muscular atrophy diagnostics[J].J Child Neurol,2007,22(8):952-956.
[5] 曲晓星,肖冰,季星,等.应用荧光定量PCR 开展上海地区脊肌萎缩症携带者的人群筛查[J].中华医学遗传学杂志,2013,30(1):1-4.
[6] Su YN,Huang CC,Lin SY,et al.Carrier screening for spinal muscular atrophy(SMA)in107,611pregnant women during the Period 2005-2009:A prospective population-based cohort study[J].PloS One,2011,6(2):e17 067.
[7] Roy N,McLean MD,Besner-Johnston A,et al.Refined physical map of the spinal muscular atrophy gene(SMA)region at 5q13based on YAC and cosmid contiguous arrays[J].Genomics,1995,26(3):451-460.
[8] 卢丽萍,麻宏伟,姜俊,等.脊髓型肌萎缩临床表型与基因拷贝数变化的相关性研究[J].中华遗传学杂志,2007,24(2):144-147.
[9] Lalic T,Vossen RH,Coffa J,et al.Deletion and duplication screening in the DMD gene using MLPA[J].Eur J Hum Genet,2005,13(11):1 231-1 234.
[10] Schouten JP,McElgunn CJ,Waaijer R,et al.Relative quantification of 40nucleic acid sequences by multiplex ligation-dependent probe amplification[J].Nucleic Acids Res,2002,30(12):e57.
[11] 孙维,沈嘉玮,龙飞,等.优选短串联重复序列应用于脊髓肌萎缩症产前诊断的连锁分析[J].上海交通大学学报,2010,30(6):707-712.
[12] Zerres K,Rudnik-Schoneborn S.Natural history in proximal spinal muscular atrophy.Clinical analysis of 445patients and suggestions for a modification of existing classifications[J].Arch Neurol,1995,52(5):518-523.
[13] Dastur RS,Gaitonde PS,Khadilkar SV,et al.Correlation between deletion patterns of SMN and NAIP genes and the clinical features of spinal muscular atrophy in Indian patients[J].Neurol India,2006,54(3):255-259.
[14] Chen Wj,Wu ZY,Wang N,et al.Quantitative studies on SMN1gene and carrier testing of Spinal muscular atrophy[J].Chin J Med Genet,2005,22(6):599-602.
猜你喜欢
临床输血与检验(2022年3期)2022-06-22
右江民族医学院学报(2022年2期)2022-05-19
河北医学(2021年10期)2021-10-27
山东医药(2021年24期)2021-09-01
诊断学(理论与实践)(2020年1期)2020-04-28
中国临床医学影像杂志(2019年6期)2019-08-27
郑州大学学报(医学版)(2019年3期)2019-06-03
中国生殖健康(2018年1期)2018-11-06
今日中国·中文版(2017年8期)2017-09-03
今日中国·中文版(2017年8期)2017-08-14
