
德朗综合征1例报告及基因分析
2017-04-26 02:50杨保旺徐菱阳王爱华
临床儿科杂志 2017年3期
杨保旺 徐菱阳 王爱华
兰州大学第二医院(甘肃兰州 730000)
德朗综合征1例报告及基因分析
杨保旺 徐菱阳 王爱华
兰州大学第二医院(甘肃兰州 730000)
目的探讨遗传代谢病德朗综合征(CdLS)的临床特点及基因突变类型。方法回顾1例CdLS患儿的临床资料及基因检测结果。结果患儿,男,2岁。外貌特殊,头小,眉毛浓密、双侧汇合于中线,睫毛长而卷曲,体质量低,运动及智力发育落后。检测患儿NIPBL基因发现c.7176T>A(编码区7176号核苷酸由T变为A)的杂核苷酸变异。结论CdLS为罕见先天遗传代谢病,临床表现有特殊的面容及体征。NIPBL基因的c.7176T>A突变在国内外未见报道。
德朗综合征; 基因突变; 遗传代谢病
德朗综合征(Cornelia de Lange syndrome,CdLS)又名阿姆斯特丹侏儒症,是一种少见的遗传病,临床可表现为多器官发育畸形,发病率为0.6/100 000[1],患儿无性别差异,同胞发病率为2%~5%。本文报告1例CdLS患儿的临床资料及基因分析结果。
1 临床资料
患儿,男,2岁10个月。因发热、咳嗽3天,加重伴精神反应差1天收住兰州大学第二医院。患儿系孤儿,出生史不详,生后送福利院。患儿生长发育落后,入院体质量仅4.8 kg、身长67 cm、头围40 cm,竖头欠稳,能翻身,不会爬,不会坐,不会说话。精神可,前囟闭合,有特殊面容,毛发浓密,前后发际低,连眉,睫毛长,鼻梁低平,鼻头小而上翘,人中长且外凸,嘴唇薄,颧弓高(图1)。眼睑无浮肿,睑结膜无苍白,巩膜无黄染,瞳孔等大等圆,直径3 mm,对光反射灵敏。颈软,无抵抗。双肺呼吸音粗,可闻及痰鸣音。心、腹检查未见异常。扁平手,通贯掌。阴茎短小,双侧阴囊空虚。双下肢膝腱反射亢进。入院后心脏彩色超声示:肺动脉瓣及瓣上狭窄(轻度),降主动脉狭窄(轻度),肺动脉高压(中度),心功能正常,三尖瓣反流(中度),肺动脉瓣反流。胸部正位片示:双侧支气管肺炎。初步诊断为支气管肺炎。因患儿有特殊面容、多器官发育畸形、体质量极低,且伴运动及智力发育落后,考虑有先天性遗传代谢性疾病,为CdLS,常见基因变异为NIPBL基因突变。
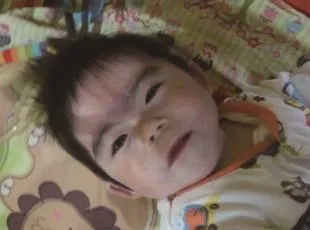
图1 患儿特殊面容
征得福利院工作人员同意后,行相关基因及染色体检查。染色体检查采用G显带方法,计数20个分裂组,核型分析5个,未发现染色体数目或结构异常。基因检测采用二代测序(高通量测序)、一代测序方法。利用FlexiGene DNA Kit提取基因组DNA,采用Nanodrop 2000超微量分光光度计(美国Thermo Fisher Scientific公司)测定DNA含量。靶向捕获利用Agilent SureSelect Target Enrichment System Kit,使用RTA software(real-time analysis,Illumina)、CASAVA software v1.8.2(Illumina)、BWA、Genome Analysis Toolkit(GATK)软件分析后,评估测序质量。运用PolyPhen-2.2.2等软件进行变异注释,最后,采用Sanger法对候选变异进行验证。基因检查结果为NIPBL基因突变:NIPBL基因c.7176T>A(编码区7176号核苷酸由T变为A)的杂核苷酸变异。该变异导致编译第2392号氨基酸Cys的密码子变为终止密码子(p.Cys2392Ter),从而使肽链合成提前终止,为无义突变。该变异可能导致蛋白质功能受影响(图2)。该变异为新发变异,其致病性尚未见文献报道(所参考数据库:HGMD Pro及PubMed),该变异不属于多态性变化,在人群中的发生频率极低。用MutationTaster软件对蛋白功能损伤预测该变异为很可能致病(disease_causing_automatic),故认为该变异为本病的致病基因,患儿CdLS基因诊断明确。

图2 患儿基因检测结果
2 讨论
CdLS是一种先天多发性发育异常综合征[2]。1933年首先由 Cornelia de lange 描述,为常染色体显性遗传性疾病。检索相关数据库(CNKI/万方/维普数据库),国内共有相关文献报道17篇,有相关病例20例,所有患儿全部具有特殊面容(一字眉/连眉,睫毛长且卷曲,毛发浓密,耳位低、鼻梁低平),通贯手,体质量及智力均较低下。主要依据临床诊断,仅有4例患儿进行了基因诊断,其中2例有基因突变,均为NIPBL基因突变。
Van Allen等[3]将CdLS分为3型。Ⅰ型即经典型,具有典型的面容及骨骼畸形,多有宫内发育迟缓,生后中到重度精神运动发育迟滞,伴有严重畸形,最终导致严重残疾或死亡;Ⅱ型即轻型,有相似面容,但骨骼畸形较经典型轻,可随年龄增长逐渐表现出来,轻度精神运动发育迟滞,很少有严重的宫内、宫外发育迟缓;Ⅲ型多与染色体的非整倍体或致畸突变有关。也有学者将CdLS分为典型及轻型两大类[4],两者均有较易分辨的特殊面容,但轻型患者极少有四肢和心脏畸形,且轻型CdLS患儿的认知能力缺陷相对较轻。本病主要临床特征包括[2]:①产前和产后发育滞后,出生体质量低,生后发育落后;②小头;③严重智力发育滞后,语言落后;④喂养困难;⑤畸形,主要为肢体残缺,表现小手、小脚,并指,第5指内弯;⑥特征面容,弓形眉、连体眉、短鼻、鼻孔前倾、长人中、耳位低、上唇薄、嘴角上翘、颚/颌小。CdLS的诊断主要根据病史及临床表现,确诊需行基因学诊断。本病无有效治疗手段。有报道如合并多系统畸形如肢体畸形、先天性心脏病、腭裂以及生后喂养困难等,影响患儿预后。
CdLS病因不详,通常认为与基因突变有关,目前已识别的CdLS致病基因有3个,分别是位于5号染色体的NIPBL基因[5](约50%患者存在该基因突变),位于X染色体上的SMCIA基因[6](约5%的患者存在该基因突变),和位于10号染色体的SMC3基因[7](目前仅有1例报道)。目前国内报道了20例CdLS患儿,进行基因突变测定的4例中2例发现突变基因,均为NIPBL基因[8],1例为NIPBL基因20号外显子检测到c.4321G>T杂合突变,另1例为NIPBL基因38号内含子c.6589+5G>C杂合突变。
本例患儿有特殊面容、扁平手、通贯掌、阴茎短小、双侧阴囊空虚,且有低体质量,运动及智力发育落后,其临床表现及特殊的外貌体征符合CdLS表现。基因检测发现有NIPBL基因c.7176T>A的杂核苷酸变异。该变异为新发现的基因突变,其致病性尚未见文献报道。用MutationTaster软件对蛋白功能损伤预测显示该变异为很可能致病。因此认为该变异为本病例的致病基因。NIPBL基因是CdLS I型的致病基因,为常染色体显性遗传。对于该类遗传方式,杂合变异可能导致发病。由于该患儿系孤儿,故未能对其父母进行基因检测。
参考文献:
[1] Liu J, Baynam G. Cornelia de Lange syndrome [J]. Adv Exp Med Biol, 2010, 685:111-123.
[2] Oliver C, Bedeschi MF, Blagowidow N, et al. Cornelia de Lange syndrome: extending the physical and psychological phenotype [J]. Am J Med Genet A, 2010, 152A(5):1127-1135.
[3] Van Allen MI, Filippi G, Siegel-Bartelt J, et al. Clinical variability within Brachmann-de Lange syndrome: a proposed classification system [J]. Am J Med Genet, 1993, 47(7):947-958.
[4] Ireland M, Donnai D, Burn J. Brachmann-de Lange syndrome. Delineation of the clinical phenotype [J]. Am J Med Genet, 1993, 47(7):959-964.
[5] Krantz ID, McCallum J, DeScipio C, et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B [J]. Nat Genet,2004, 36(6):631-635.
[6] Musio A, Selicorni A, Focarelli ML, et al. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations [J]. Nat Genet, 2006, 38(5):528-530.
[7] Kalal GI, Raina VP, Nayak VS, et al. Cornelia de Lange syndrome: a case study [J]. Genet Test Mol Biomarkers, 2009, 13(1):15-18.
[8] 钟秋连. 运用细胞和分子遗传学方法对Cornelia de Lange综合征患者进行遗传诊断 [D]. 中南大学, 2011.
Genetic analysis of Cornelia de Lange syndrome in one case
YANG Baowang, XU Lingyang, WANG Aihua
(The Second Hospital of Lanzhou University, Lanzhou 730000, Gansu, China)
ObjectiveTo explore the clinical features and gene mutation types of Cornelia de Lange syndrome (CdLS), an inherited metabolic disease.MethodsThe clinical data and gene detection results of one case of CdLS were retrospectively analyzed.ResultsTwo-year-old male had special appearance, microcephaly, bushy eyebrows with both sides meeting in the midline, long curly eyelashes, low body mass, and motor and mental retardation. NIPBL gene detection found the variation of the nucleotide in c.7176T>A (nucleotide 7176 in coding region changed from T to A).ConclusionsCdLS is a rare congenital inherited metabolic disease. The clinical manifestations were special appearance and signs. The c.7176T>A mutation in NIPBL gene has not been reported at home and abroad.
Cornelia de Lange syndrome; gene mutation; inherited metabolic diseases
10.3969/j.issn.1000-3606.2017.03.013
2016-08-29)
(本文编辑:蔡虹蔚)
王爱华 电子信箱:aihua970@163.com
猜你喜欢
风流一代·青春(2021年9期)2021-09-23
中国生殖健康(2020年2期)2021-01-18
科学之谜(2019年3期)2019-03-28
幸福·婚姻版(2018年9期)2018-11-26
科学之谜(2018年8期)2018-09-29
小学生导刊(2018年13期)2018-06-29
草原(2018年2期)2018-03-02
中国生殖健康(2018年2期)2018-01-12
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
