
六聚吡咯大环与铀/超铀酰基离子相互作用:成键、热力学和光谱性质
2018-06-06 05:50毕艳婷沈中辉张红星潘清江
无机化学学报 2018年6期
毕艳婷 姚 军 沈中辉*, 张红星 潘清江*,
(1黑龙江大学科学技术处,化学化工与材料学院,哈尔滨 150080)(2吉林大学理论化学研究所,长春 130023)
铀和超铀元素(An)是生产核能、制造核武器和同位素药物的重要原料[1]。由于具有高毒性、高放射性,在核反应堆操作运行、乏燃料回收处理、核废料长期处置以及材料运输过程中产生的泄漏已引发诸多环境问题[2-4]。目前,采用适当配体、进行超低浓度条件下的传感检测能够在一定程度上防患于未然;而使用合适配体分离回收乏燃料和核废料中高放射性锕系元素及其衍生物则是解决核素污染的有效手段之一[3]。
相对镧系元素的收缩4f轨道特征(core-like),锕系的5f轨道在形成配合物时可参与成键[5-13]。锕系元素能与元素周期表中很多元素形成多种类型化学键。对于六价高氧化态的U、Np和Pu,通常以线性反式锕酰基离子结构形式存在[1]。如trans-UO22+具有高热力学稳定性和强动力学惰性,广泛存在于核燃料和核废料中;其易溶于水,能够在自然水体系中迁移,进而被生物体吸收、造成极大危害。由于AnO22+轴向An=O化学键很强、表现出一定化学惰性,因而其赤道平面方向的配位化学研究相当活跃。当前,除含O和P供体原子的配体外,N-供体配体已逐渐在锕系元素检测和分离中扮演越来越重要的角色[3,14-23]。如Sessler教授课题组报道只需观察溶液颜色变化,就可采用Hexaphyrin配体检测锕酰离子;结构表征发现配体空穴很好地容纳锕酰离子[3,14-20,36]。
为进一步开发具有高效分离特性、高选择性的含N配体,它与锕系元素相互作用行为、成键性质、电子结构本质和化学反应行为方面研究是非常迫切的。由于锕系元素本身的稀缺性、高毒性、高放射性给实验研究带来诸多困难,这为基于准确方法论的理论计算提供了契机与挑战。本文中,设计3种六聚吡咯大环(标记为 H4Ln,n=1~3;Scheme 1)和一种实验已合成配体(H4L0)。采用相对论密度泛函理论探索形成的铀和超铀配合物结构和成键性质,将通过热力学反应计算和光谱性质分析得到金属与配体的相互作用信息。
1 计算方法和细节
计算系列六聚吡咯大环与锕酰基离子形成的配合物[(AnO2)(Ln)]2-,标记为 nAn(其中 n=1~3,An=U、Np和Pu)。为便于对比,还优化了实验已经合成的配合物[(UO2)(L0)]2-(A)[14],其中L0具有6个甲基和6个乙基取代基团(Scheme 1)。

Scheme 1 Theoretically designed L1,L2and L3,compared with experimentally synthesized L0
结构优化采用Priroda[24]程序完成。使用全电子标量哈密尔顿方法、全电子高斯基组以及广义梯度近似PBE泛函。解析频率计算未得到虚频,表明该结构为势能面的极小值稳定点。频率计算得到吉布斯自由能等热力学参数,同时还根据频率值和红外活性强度拟合红外振动光谱。计算同时获得配合物Mayer键级。
使用ADF2014程序计算配合物体系溶剂化能。积分格点选用6.0×6.0×6.0。溶剂化效应采用COSMO模型[25],溶剂水的介电常数为78.5。使用Klamt半径,H 0.130 nm、C 0.200 nm、N 0.183 nm、O 0.172 nm和An 0.170 nm[25-28]。应用标量相对论ZORA方法、Slater型TZP基组和PBE泛函进行计算。
使用Gaussian09程序[29]计算配合物电子吸收光谱。采用含时密度泛函理论(Time-dependent density functional theory,TD-DFT)方法,运用杂化 B3LYP 泛函,计算得到配合物的自旋允许的80个低能激发态。对金属铀使用Stuttgart准相对论有效核势和相应赝势基组,对其它原子使用6-31G**基组。采用CPCM模型模拟溶液水的环境效应。此外,对配合物进行QTAIM(Quantum Theory of Atoms in Molecule)[30]拓扑分析,结合Multiwfn软件[31]获得在An-N临界点(Bond critical points,BCPs)的电子密度 ρ(r)、拉普拉斯密度▽2ρ(r)、能量密度H(r)、椭圆率 ε 和相关作用能Eint,以及Mayer和Wiberg键级。
2 结果与讨论
2.1 几何结构和振动光谱
对于吡咯环位置不同的L1和L2异构体,计算发现配合物1U要比2U能量高77 kJ·mol-1。它们的铀酰基团保持近似线性结构 (177°和180°),见表1和图 1;1U 的 N-U-N 角数值从 57.9°到 62.5°(平均值为59.9°),2U的在相近范围内。优化的U=O键长均为0.180 nm,这一数值在实验[3,14-18,32]和计算[5-6]配合物的U=O距离范围内。优化得到A的U=O键长也是0.180 nm,比其实验值0.176 nm略长[14]。这与GGA泛函高估键长相关[33]。计算的1U的U-N键长范围为0.261~0.293 nm,而2U的U-N键长0.260~0.291 nm,后者的略短。就结构而言,这一稍短U-N距离可能是2U相对1U稳定的原因。类似地,An-N键长的比较也可以近似判断同分异构体1Np和2Np以及1Pu和2Pu间的相对稳定性。
配体不变,按U、Np、Pu的次序变化,1An的An=O 键长依次为 0.179 9、0.181 6、0.179 7 nm,An-N平均值为0.281、0.282、0.286 nm,并未直接反应出锕系收缩这一原理。然而,计算的An=O键级分别为2.39、2.38、2.37,表明其键强度随着元素原子序数增加而逐渐减小。这一结构特征将在红外振动光谱分析时给出进一步证据。当金属中心不变,配体变化时,得到1U的N-U-N键角总和为360°,反映六聚吡咯配体中所有氮原子几乎位于同一个赤道平面上,同时表明L1所形成空穴尺寸与金属中心离子大小相匹配。因为L1和L2是异构体,它们空穴应该相当,而计算的2U的360°键角总和也证明这一点。因而,得到的1U和2U中,铀酰基离子的空间构型为较规则的六角双锥结构。计算A的N-U-N键角总和为366°,吡咯氮原子略偏离赤道平面,这表明周边取代基对配体空穴尺寸有少量影响。有趣的是这一数值与实验数值完全吻合[14]。相比之下,3U的键角总和为376°,L3的6个吡咯氮原子偏离赤道平面,中心离子与配体空穴尺寸不匹配,因而它采用扭曲六角双锥结构,以降低体系能量、稳定配合物。

图1 配合物nAn(n=1~3;An=U、Np和Pu)和参考分子A的优化结构Fig.1 Optimized structures of complexes nAn(n=1~3;An=U,Np and Pu)and experimentally synthesized A
频率计算得到nU在900~910 cm-1范围内有强U=O伸缩振动吸收峰(图S1左图)。计算的A的U=O振动吸收峰在904 cm-1处,与实验报道的922 cm-1相对应[14-16];由于计算采用GGA泛函,其泛函本质导致稍微低估键的强度,即会低估对应的伸缩振动频率数值[33]。同时,计算的U=O数值在已报道的铀酰基配合物的振动吸收峰范围内[3,14-18,32]。当配体相同时,An=O伸缩振动频率按U、Np、Pu顺序向低频移动,即发生红移现象。如1An分别在910、887、874 cm-1处有强的An=O伸缩振动吸收 (图S1右图)。这是锕系收缩规律在红外光谱中的体现。这与结构计算的An=O键级变化趋势一致。同样地,该规律也适用于2An和3An,见补充材料中图S2。中心离子相同、配体变化对配合物的An=O振动吸收频率有一定的影响,且具有金属种类依赖性。
2.2 配体和锕系元素相互作用的拓扑分析
为进一步理解配体与锕系离子相互作用本质,计算得到 1U、2U、3U、1Np和 1Pu在 An-N键临界点的 ρ(r)、▽2ρ(r)和 H(r)以及 ε 等 QTAIM 参数。 平均数值列于表2中,每一个An-N键的详细数据见表S4。以往报道证明,基于电子密度的QTAIM拓扑分析能很好地表征供体配体与锕系元素之间形成化学键的性质和强度[7-9,30],并能对传统的波函数分子轨道解释给出有力补充。一般地,在化学键临界点的H(r)小于或接近零时,表明共价键性质,绝对值大小表示共价键强弱的程度;ρ(r)<0.10 a.u.、▽2ρ(r)>0 时为闭壳层作用,包括离子键、范德华作用或氢键等;ρ(r)>0.20 a.u.、▽2ρ(r)<0 为开壳层作用或者共价键。
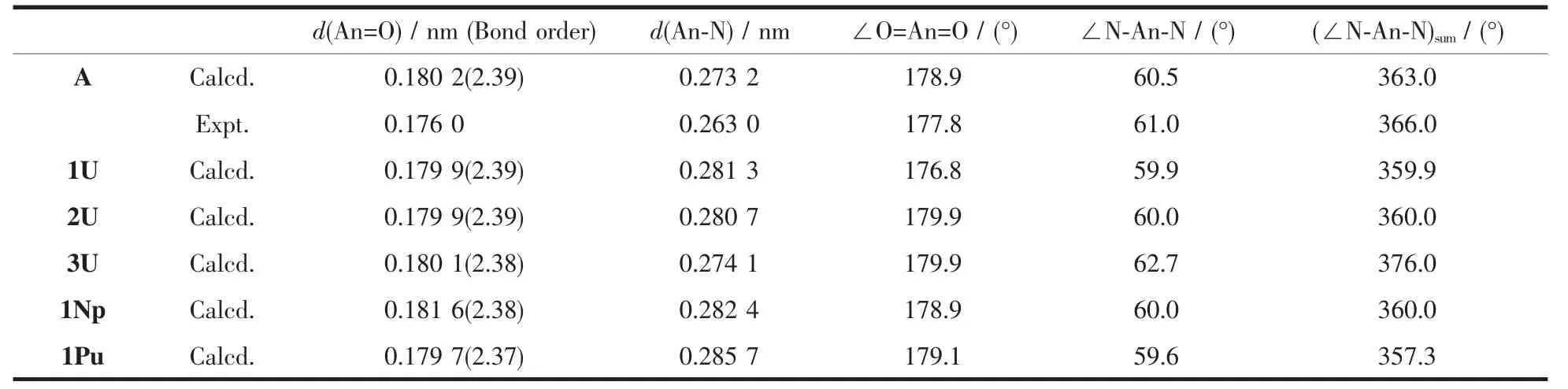
表1 配合物A和nAn(An=U、Np和Pu)的优化平均键参数以及参考配合物A的实验值Table 1 Optimized average bond parameters for A and nAn (An=U,Np and Pu)compared with experimental values of A
检查表2中的QTAIM参数可以看到,计算的含铀配合物的H(r)均为负值,绝对值数值较小说明U-N为强度很弱的共价键性质。这些铀配合物的电子密度 ρ(r)数值很小,在 0.033 2~0.036 1之间,且▽2ρ(r)均为正值(0.087~0.110),表明 U-N 键中有很大的离子性质。在U-N临界点的椭圆率ε可以度量化学键的圆柱形对称性,计算结果表明U-N为单键特征。因此,我们指认U-N为具有大部分离子性的弱共价键。此外,同分异构体1U和2U的QTAIM参数数值几乎完全相同,表明吡咯单元的联接方式对金属离子和配体供体之间作用影响极其微小。对于设计的配合物3U,由于结构联接使得配体具有相对的柔性,即配体可以通过一定程度的扭曲来加强配体和金属铀的相互作用,以达到降低体系能量、稳定体系的目的,因而,其U-N临界点处的ρ(r)数值相对1U和2U的略有增大,即U-N键强度略有增加。显然,QTAIM分析得到的这一结论与前文对几何参数U-N键长分析结果相一致。相对1U,配合物A在配体周边增加了6个甲基和6个乙基基团,其推电子作用会增强吡咯氮对金属中心的供体作用,因此,后者具有相对大些的电子密度数值。
分析比较3个1An配合物的电子密度和能量密度数据,可以看出它们的ρ(r)数值按U、Np、Pu的次序逐渐变小,表明它们的An-N作用逐渐变弱。1Np和1Pu的H(r)虽然为正值,但是仍非常接近零,因此我们仍指认Np/Pu-N为离子性为主、强度较弱的共价键性质。能量密度H(r)可分解为动能项G(r)和势能项V(r),其中V(r)的一半可表示为An-N的相互作用能Eint(kJ·mol-1)。如表2所示,当金属中心为铀,配体按 L1→L2→L3递变,Eint分别为-30.96、-31.38 和-35.15 kJ·mol-1, 即数值逐渐变小,N→U配位作用增强;这与配合物1U(0.281 3 nm)、2U(0.280 7 nm)和3U(0.274 1 nm)的U-N平均键长变化趋势相一致。比较1An,随着U→Np→Pu变化,其Eint数值逐渐增大、配位作用减弱,这与其An-N键长逐渐增长密切相关(表1)。另外,计算得到配合物的QTAIM数据与An-N的键级有一定的相关性,特别是Priroda计算得到的Mayer键级与电子密度ρ(r)变化趋势一致。例如,随着1U→3U和1U→1Np→1Pu的ρ(r)数值增加,此Mayer键级表现出同样增加的趋势。然而,QTAIM计算的Mayer和Wiberg键级变化趋势与电子密度有所差异。这一现象在以前的QTAIM计算中也有报道[7,34-35],因此在使用该键级数据时要谨慎。

表2 配合物在An-N键临界点处的QTAIM参数平均值、ε、Eint以及各种方法计算的键级Table 2 Average QTAIM parameters of the complexes at An-N bond critical points including electron density (ρ(r)),Laplacian (▽2ρ(r)),energy density (H(r)),ellipticity (ε)and interaction energy(Eint),together with bond orders calculated by different approaches
2.3 热力学反应
选择多种锕源前体,设计与六聚吡咯配体的反应(表3),并计算了在溶液介质环境下的反应自由能ΔrG(aq)列于表4。更为详细的能量数值,包括气态条件下的总反应能ΔrE(gas)、含零点振动能校正的总能ΔrE0(gas)、自由能ΔrG(gas)以及溶剂效应影响ΔrG(aq),列于表S5~S7。计算表明介质环境对反应能影响很大,ΔrG(aq)数值超过 1 255 kJ·mol-1,因此研究中必须考虑溶液环境的影响。对于反应(3),配合物中心离子相同时,按配体 L2、L1、L3顺序,溶液条件下反应自由能ΔrG(aq)依次增大;其变化趋势如图2所示。如铀配合物2U、1U和3U反应自由能分别为-90、-56 和 46 kJ·mol-1。就热力学而言,前 2 个配合物相对容易生成。这与实验发现配体L0可以用于锕酰基离子检测的结果相吻合[14-16]。同时还发现,当配合物中配体相同,按 U、Pu、Np 的顺序,ΔrG(aq)依次减小,如以1An为例,反应自由能依次为-56、-108、-117 kJ·mol-1,表明反应越来越容易。实验测量L0探测不同锕酰基离子发现,探测铀酰需要相对较长时间,一般数个小时后会发现检测前后物质的颜色有所变化,也就是反应相对较慢;而超铀酰基则是很短的时间内发生配位反应,观察到颜色变化[14-16]。从热力学能量角度出发,以上计算的1An的ΔrG(aq)相对数值大小验证了实验观察。同样的规律发生在反应(1)和(2)中。
由于配合物A已经实验合成并进行结构和性质表征,因此,其参与反应所获得的相关参数可作为评价其它配合物是否可以形成或作为传感配体的参考依据。计算得到A在反应(1)~(3)中的ΔrG(aq)分别为147、110和101 kJ·mol-1。显然设计的配合物多数都小于这一数值(图2)。只有在反应(2)中,3U的自由能比A的大~25 kJ·mol-1。总之,配合物nAn相对较小的ΔrG(aq)数值(大部分为负值)说明它们可以在相对温和实验条件下获得,相应的大环配体有可能应用于分离锕系离子。

表3 配合物的生成反应Table 3 Formation reactions of actinyl complexes of polypyrrolic macrocycles

表4 计算的配合物A和1An(An=U、Np和Pu)在水溶液中反应自由能Table 4 Calculated reaction free energies ΔrG(aq)of A and 1An (An=U,Np and Pu)in aqueous solution kJ·mol-1
2.4 电子吸收光谱
采用TD-B3LYP方法计算4个铀配合物在水溶液中的电子吸收,并采用Gaussian函数展开拟合了光谱(图 3)。1U 的吸收波长(λ)、电子跃迁能(ΔE)和振子强度(f)列于表5中,而包括组态跃迁等详细信息见表 S8~S10。
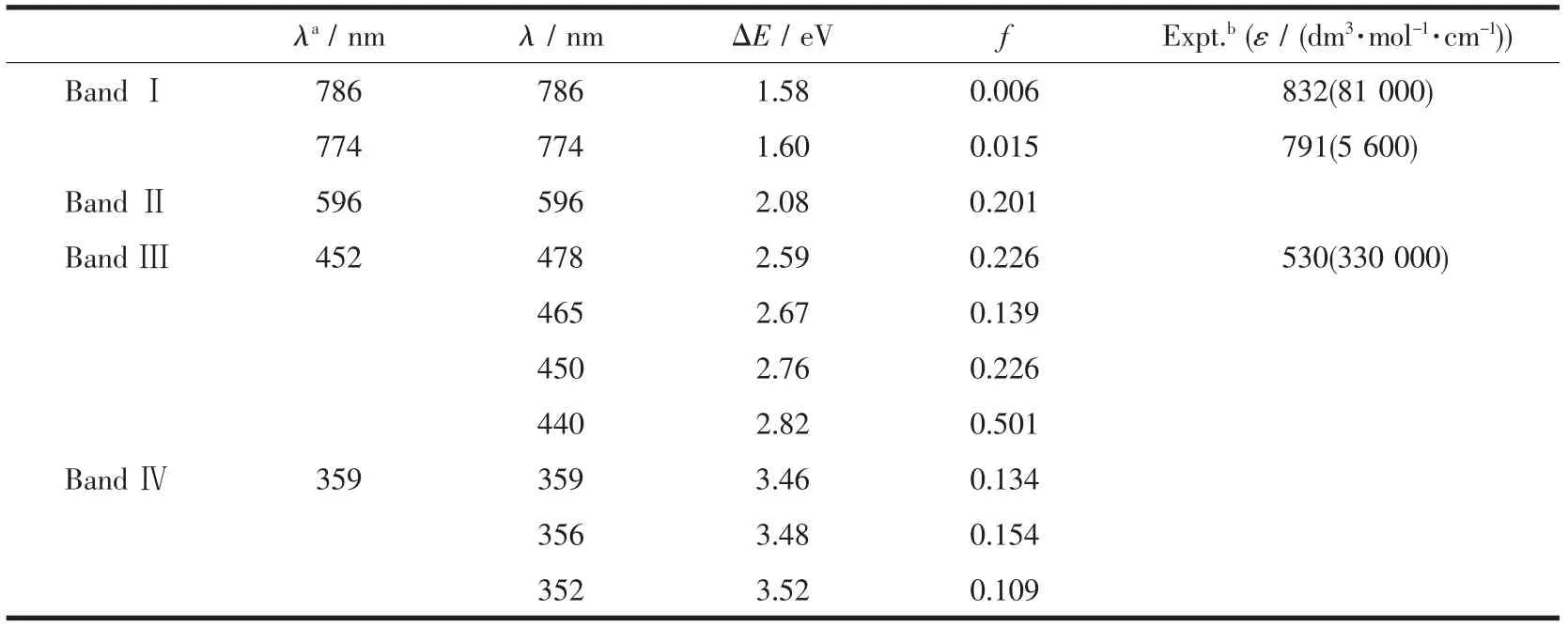
表5 TD-B3LYP计算的1U吸收波长(λ)、电子跃迁能(ΔE)和振子强度(f)Table 5 Absorption wavelength (λ),excitation energy (ΔE)and oscillator strength (f)of 1U in the aqueous solution calculated at the TD-B3LYP level
首先讨论实验合成配合物A的电子吸收光谱。在低能近红外区域计算得到2个振子强度相对较小的吸收峰821和805 nm。这一计算结果与实验得到的832和791 nm的吸收峰相接近[14-16]。激发态波函数分析表明,它们为配体π(L0)到金属中心U(5f)的电子跃迁 (ligand-to-metal charge transfer,LMCT),同时混合很少量的 π(L0)→π*(L0)跃迁(intraligand charge transfer,ILCT)。需要指出的是,我们计算了A的80个自旋允许的低能激发态,仅得到其在近红外区域的光谱,如需要可见光和紫外区光谱跃迁,则需要考虑更多的激发态。
1U和2U是同分异构体,差异只是吡咯环连接位置不同,因此计算得到相似的电子吸收光谱 (图3)。1U 在 786 nm(1.58 eV)和 774 nm(1.60 eV)处有 2个很弱吸收峰,它们分别是HOMO-1→LUMO和HOMO-2→LUMO两个跃迁组态贡献,CI组合系数为0.694和0.701。轨道分析(图S3)指认为LMCT性质跃迁。在可见光区域,计算得到1U有2个吸收峰,分别出现在596和452 nm处,前者为π(L1)→π*(L1),并混有少量 δ(U(5f))→π*(L1)性质;而 452 nm吸收是LMCT和ILCT两种跃迁并重性质。在低能区339 nm处,1U具有π(L1)→π*(L1)性质的强吸收光谱带。可以看出,配合物1U的电子吸收光谱与实验得到的 832 nm(ε=8.1×104dm3·mol-1·cm-1)、791 nm(ε=5.6×103dm3·mol-1·cm-1)和 530 nm(ε=3.3×105dm3·mol-1·cm-1)相对应。 由于 1U 中未包括周边的甲基等取代基,使得其波长数值相对实验测定的短,但是它们吸收峰的强度有很好的对应关系。同样地,2U的吸收光谱包括4个吸收带,即低能近红外区的800和767 nm(BandⅠ)、可见光区域的594 nm(BandⅡ)和431 nm(BandⅢ)、以及高能紫外区的344 nm(BandⅣ)吸收峰,且它们的跃迁性质与1U的一致。当然,配体的细微差别体现在它们的吸收峰波长略有差异。
从图3可以看出,3U的吸收光谱与1U和2U大致相同,如具有上述的4个主要特征带。然而含更多亚甲基的L3配体使得3U表现出一些不同的吸收特征。如近红外区的2个吸收峰相距增大、可见光区域的相对低能的607 nm峰强度相对变弱,另外在557 nm处出现了独有的吸收肩峰。波函数分析不难看出,在1U和2U中,配体具有刚性的共轭效应、结构区域平面,这不同于扭曲的L3配体。
3 结 论
采用相对论密度泛函理论探索10种铀/超铀配合物的成键、热力学反应和光谱性质。结构优化表明,配合物1An和2An配体空穴尺寸与金属中心原子大小较匹配,六聚吡咯配体中的6个氮供体原子与An位于同一赤道平面上;配合物3An的配体空穴与金属中心原子大小匹配性较差,吡咯氮原子偏离赤道平面。当配体相同,按铀、镎、钚顺序An=O振动频率向低频移动,这与周期表“锕系收缩”规律相一致。同时,与计算的An=O的键长和键级变化规律是一致的。QTAIM拓扑分析发现An-N键为离子性占主要成分的弱共价键。热力学反应能计算表明,对比实验合成配合物A的反应自由能,所设计的系列配合物均能在较温和的实验条件下生成,且溶剂化效应对反应影响很大。在电子吸收光谱研究中发现,nU和A在低能近红外和可见光区域出现LMCT和ILCT的混合跃迁性质吸收带,与实验测定的吸收光谱符合。与大环配体吸收光谱的比较表明该类配体有作为锕系离子检测传感器应用的前景。本文研究一方面可为新型结构锕系配合物设计提供相应的理论支持;另一方面,计算得到的配合物电子结构、光谱性质、配体与锕系离子相互作用以及反应能等信息也可为铀和超铀元素的提纯和分离提供理论参考。
Supporting information is available at http://www.wjhxxb.cn
[1]Hashke J M,Stakebake J L.The Chemistry of the Actinide and Transactinide Elements.Dordrecht:Springer,2006:3199-3272
[2]Choppin G R.J.Radioanal.Nucl.Chem.,2007,273(3):695-703
[3]Rambo B M,Sessler J L.Chem.-Eur.J.,2011,17(18):4946-4959
[4]GU Jia-Fang(辜家芳),XU Ke(许可),CHEN Wen-Kai(陈文凯).Chinese J.Inorg.Chem.(无机化学学报),2017,33(9):1579-1586
[5]Wang D,van Gunsteren W F,Chai Z.Chem.Soc.Rev.,2012,41(17):5836-5865
[6]Wang D,Su J,Wu J,et al.Radiochim.Acta,2014,102(1/2):13-25
[7]Wu Q Y,Wang C Z,Lan J H,et al.Inorg.Chem.,2014,53(18):9607-9614
[8]Wu H,Wu Q Y,Wang C Z,et al.Dalton Trans.,2015,44(38):16737-16745
[9]Lan J H,Shi W Q,Yuan L Y.Coord.Chem.Rev.,2012,256(13/14):1406-1417
[10]Lan J H,Shi W Q,Yuan L Y,et al.Inorg.Chem.,2011,50(19):9230-9237
[11]Hu H S,Wei F,Wang X,et al.J.Am.Chem.Soc.,2014,136(4):1427-1437
[12]Hu S X,Jian J,Su J,et al.Chem.Sci.,2017,8(5):4035-4043
[13]Chi C,Wang J Q,Qu H,et al.Angew.Chem.Int.Ed.,2017,56(24):6932-6936
[14]Sessler J L,Seidel D,Vivian A E,et al.Angew.Chem.Int.Ed.,2001,40(3):591-594
[15]Sessler J L,Gorden A E V,Seidel D,et al.Inorg.Chim.Acta,2002,341:54-70
[16]Sessler J L,Melfi P J,Seidel D,et al.Tetrahedron,2004,60(49):11089-11097
[17]Melfi P J,Kim S K,Lee J T,et al.Inorg.Chem.,2007,46(13):5143-5145
[18]Ho I T,Zhang Z,Ishida M,et al.J.Am.Chem.Soc.,2014,136(11):4281-4286
[19]Zhang Z,Dong S K,Lin C Y,et al.J.Am.Chem.Soc.,2015,137(24):7769-7774
[20]Sessler J L,Melfi P J,Lynch V M.J.Porphyrins Phthalocyanines,2007,11(4):287-293
[21]Cafeo G,Kohnke F H,La Torre G L,et al.Angew.Chem.Int.Ed.,2000,39(8):1496-1498
[22]Jana A,Ishida M,Cho K,et al.Chem.Commun.,2013,49(79):8937-8939
[23]Fukuzumi S,Ohkubo K,D′Souza F,et al.Chem.Commun.,2012,48(79):9801-9815
[24]Laikov D N.J.Comput.Chem.,2007,28(3):698-702
[25]Klamt A,Jonas V,Burger T,et al.J.Phys.Chem.A,1998,102(26):5074-5085
[26]Zhao H B,Zheng M,Schreckenbach G,et al.Inorg.Chem.,2017,56(5):2763-2776
[27]CHEN Fang-Yuan(陈方园),QU Ning(曲宁),WU Qun-Yan(吴群 燕),et al.Acta Chim.Sinica(化学学报),2017,75(5):457-463
[28]ZHAO Si-Wei(赵思魏),ZHONG Yu-Xi(钟宇曦),GUO Yuan-Ru(郭元茹),et al.Acta Chim.Sinica(化学学报),2016,74(8):683-688
[29]Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian09,Gaussian,Inc.,Wallingford CT,2009.
[30]Bader R F W.J.Phys.Chem.A,1998,102:7314-7323
[31]Lu T,Chen F.J.Comput.Chem.,2012,33(5):580-592
[32]Natrajan L,Burdet F,Pecaut J,et al.J.Am.Chem.Soc.,2006,128(22):7152-7153
[33]Perdew J P,Burke K,Ernzerhof M.Phys.Rev.Lett.,1996,77(18):3865-3868
[34]Hlina J A,Pankhurst J R,Kaltsoyannis N,et al.J.Am.Chem.Soc.,2016,138(10):3333-3345
[35]Mountain A R,Kaltsoyannis N.Dalton Trans.,2013,42(37):13477-13486
[36]GU Jia-Fang(辜家芳),MAN Mei-Ling(满梅玲),LU Chun-Hai(陆春海),et al.Chinese J.Inorg.Chem.(无机化学学报),2012,28(7):1324-1332
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30
化学与粘合(2020年4期)2020-09-11
生物工程学报(2020年1期)2020-03-12
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中国资源综合利用(2017年1期)2018-01-22
山东工业技术(2016年15期)2016-12-01
中国粮油学报(2016年5期)2016-01-23
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
郑州大学学报(理学版)(2014年3期)2014-03-01
