
CRISPR/Cas9基因组编辑技术及其在链霉菌中的应用
2018-06-07 02:30乔龙亮庞建虎党晨阳黄海龙朱鹏
生物技术通报 2018年5期
乔龙亮 庞建虎 党晨阳 黄海龙 朱鹏
(宁波大学海洋学院,宁波 315211)
链霉菌(Streptomyces)是革兰氏阳性放线菌,能产生大量具有重要价值的天然代谢产物,在工业生产和医疗方面有着重要地位。目前临床应用的抗生素约2/3来源于链霉菌属[1],如杀虫药(阿维菌素和米尔贝霉素),抗菌素(红霉素和万古霉素),此外,该属菌株还可以产生抗肿瘤药(柔红霉素和博来霉素)及免疫抑制剂(FK506、FK520和雷帕霉素)等。目前对链霉菌代谢产物的研究多集中在抗生素领域,由于人类对抗生素的长期使用,从而出现大量多药耐药菌,为解决此问题,开发新的有效抗生素已成为当务之急[2-3]。链霉菌的全基因组测序显示链霉菌中存在许多隐性次级代谢产物生物合成基因簇(Cryptic secondary metabolite biosynthetic gene cluster,CSMG),在实验室条件下,它们多数处于沉默状态或者很低的表达水平,通过有效的分子操作方法来激活CSMG获得新抗生素类型已成为一个重要的研究方向[4]。
与模型菌株(如大肠杆菌和酿酒酵母)相比,链霉菌的遗传操作更加困难,部分原因是它们的基因组是目前原核生物中最大的,且基因组中GC含量极高。目前在链霉菌的遗传操作是十分有限的,主要分为有痕敲除方法和无痕敲除方法。早在2003年,Gust课题组[5]首次将λ-red重组系统应用于构建链霉菌敲除载体,但得到的目的菌株染色体上含有抗性筛选标记,不利于进行连续敲除。此后基于无痕敲除理念发展的位点特异性重组系统已经成为高通量遗传分析中有利和高效的手段,并得到广泛应用。目前在链霉菌中已得到应用的位点特异性重组系统有Cre/loxp系统、Dre/rox系统、Flp/FRT系统和Int-Xis系统(表1),但这些传统的基因编辑手段的缺点也十分明显,如效率低、实验周期长以及操作步骤繁琐等。而近年来,素有“魔剪”之称的规律成簇的间隔短回文重复(Clustered regularly interspaced short palindromic repeats,CRISPR)/Cas9(CRISPR-associated protein 9)系统因其不具有上述所有缺点,迅速席卷全球实验室,且被Science杂志评为2013年度最重要的科学突破之一。

表1 目前应用于链霉菌基因组编辑的方法[6-10]
一个典型的CRISPR-Cas9基因座结构分为3部分(图1):3'端是由多个重复序列(Repeat)与非重复间隔序列(Spacer)构成的CRISPR基因座;5'端是可转录为反式激活crRNA(Trans-activating crRNA,tracrRNA)的区域;中间部分由成簇的Cas基因组成,主要包括cas1、cas2和cas9。cas9编码蛋白具有核酸酶、解旋酶和聚合酶等功能。CRISPR/Cas9作为细菌中的适应性免疫机制,经人为改造后,可在细胞中实现高度灵活且特异的基因组编辑。该系统通过设计特异性向导RNA(Single guide RNA,sgRNA)序列与靶序列碱基配对,从而引导Cas9蛋白结合到靶序列处,行使DNA切割功能,再利用细胞的非同源性末端连接(Non-homologous end joining,NHEJ)或同源重组(Homologous recombina-tion,HR)修复机制对断裂的DNA进行插入缺失(Indel)、修复(Repair)或替换(Replacement)(图2)。此外,当RuvC结构域和HNH结构域同时处于失活状态时(D10A&H840A),Cas9将不具有核酸酶活性,成为dCas9(dead Cas9)[11],但仍能作为基因编辑工具,达到沉默基因效果。本文通过比较常规基因编辑技术和CRISPR/Cas9系统,说明CRISPR/Cas9系统在链霉菌中应用的优势,综述近年来CRISPR/Cas9系统在链霉菌中的应用,并对其应用潜力进行梳理,以期为相关领域的工作提供参考。
1 CRISPR/Cas9系统在链霉菌中应用的优势
CRISPR/Cas9系统从细菌和古细菌的天然免疫系统转变为定向基因编辑技术后,因其快速、高效、廉价和操作简单等优点使之迅速成为生命科学研究的热点工具。目前在链霉菌的常规遗传操作主要分为有痕敲除方法和无痕敲除方法。

图1 三种不同CRISPR/Cas9亚型的基因结构
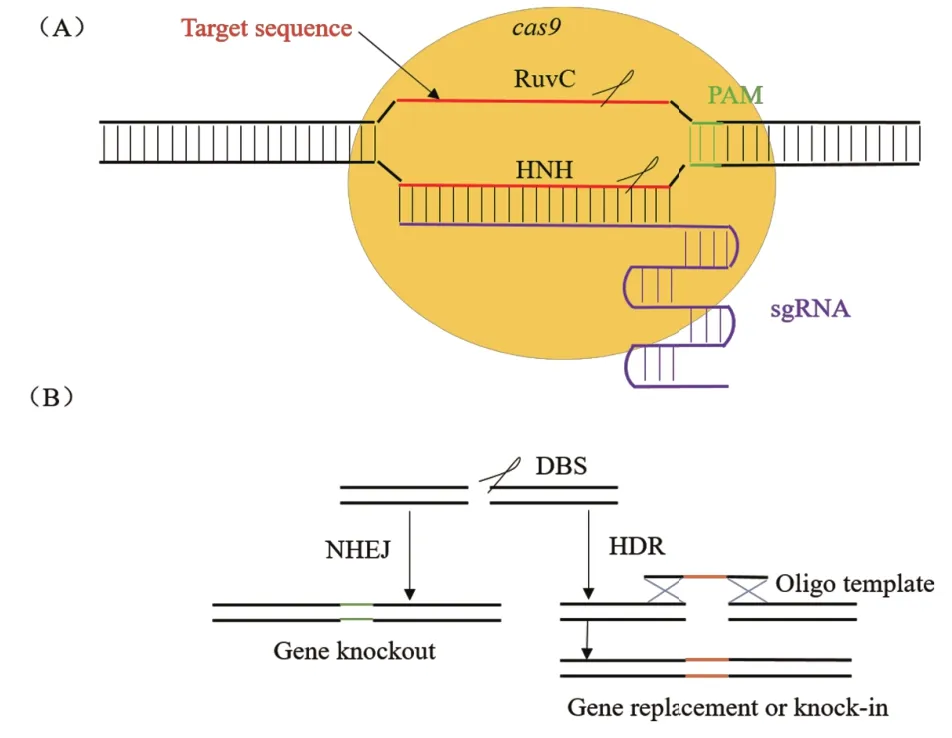
图2 CRISPR/Cas9系统介导基因编辑示意
对于有痕敲除方法,其缺点比较突出,每次敲除基因之后都会在染色体基因组上引入一个筛选标记,不利于进行连续敲除,而且抗性基因的插入还会影响靶基因下游基因的表达[12]。基于无痕敲除理念发展的位点特异性重组系统包括:Cre/loxp系统、Flp/FRT系统、Dre/rox系统和Int/Xis系统(表1)。它们在链霉菌中的应用各有千秋,但缺点也都类似,其中Cre/loxp系统、Dre/rox系统和Int/Xis系统每进行一次敲除染色体上会留下1个位点(loxp、rox、att1),连续进行多次敲除后会造成染色体的不稳定,因其步骤繁琐,实验周期长达6周。Flp/FRT系统由于flp基因GC含量太低,无法在链霉菌中表达,Gust等[5]将Flp-FRT系统和λ-red重组系统结合组成为PCR-Targeting方法对链霉菌的基因进行敲除,但效率只有40%[13]。另外,在实验周期方面至少需要花费3周时间。使用内切核酸酶I-SceI可以实现体内大片段基因敲除,但该方法尚需较长周期才能完成。而使用CRISPR/Cas9系统可以极大地简化实验步骤,实验周期缩短至17 d[14]。另外,CRISPR/Cas9系统既能敲除小的单个基因,也能敲除大的基因片段,如NRPS或PKS的整个基因簇,甚至能同时敲除多个基因,而且具有较高的效率和准确度,此前报道的其他方法是无法实现的[15]。
2 CRISPR/Cas9系统在链霉菌中的应用
模式生物因其基因组已被测序且代谢通路研究较为清晰,可以为研究CRISPR/Cas9的编辑作用以及效率提供很好的样本。目前,利用CRISPR/Cas9系统在几乎所有的模式生物中实现基因的定点敲除和编辑,在链霉菌中也有6种,分别为变铅青链霉菌(Streptomyces lividans)、绿色产色链霉菌(Streptomyces.viridochromogenes), 白 色 链 霉 菌(Streptomyces albus)、天蓝色链霉菌[Streptomyces coelicolor A3(2)]、天 蓝 色 链 霉 菌(Streptomyces coelicolorM145)、玫 瑰 孢 链 霉 菌(Streptomyces roseosporusNRRL15998),其中较为广泛研究的是天蓝色链霉菌(Streptomyces coelicolor)。普遍利用CRISPR/Cas9系统在链霉菌中对目的基因的敲除策略如图3,将根据靶基因设计的sgRNA和cas9基因以及靶基因的上下游同源臂整合在同一质粒中,转入链霉菌体内,利用转录的sgRNA引导cas9蛋白对靶基因进行切割,随后与上下游同源臂进行交换完成基因敲除。值得注意的是,由于化脓性链球菌cas9基因存在几种稀有密码子,其中有许多bldA密码子,bldA编码链霉菌中能有效识别稀有UUA亮氨酸密码的tRNA,该tRNA通过控制稀有密码子UUA的翻译来调节基因的表达[16],除了bldAtRNA以外,UUA密码子不能被其他的tRNA有效翻译。因此,在链霉菌中应用的cas9基因都需经过密码子优化。
2.1 CRISPR/Cas9系统在链霉菌中的验证

图3 敲除目的基因簇的策略
2.1.1 CRISPR/Cas9系统在链霉菌中的验证 为了验证CRISPR/Cas9系统在链霉菌中具有编辑作用,2014年,Cobb等[15]首次将CRISPR/Cas9系统应用于链霉菌。他们构建两种不同的质粒,pCRISPomyces-1质粒包括tracrRNA(由rpsLp(CF)驱动)、CRISPR阵列表达盒(由gapdhp(EL)驱动)及cas9基因;pCRISPomyces-2质粒包括sgRNA表达盒(由gapdhp(EL)驱动)和cas9基因,两种质粒中cas9基因均由启动子rpsLp(XC)-BbsI驱动。为验证两种质粒在链霉菌中行使编辑功能的效果,以变铅青链霉菌为研究对象,针对十一烷基灵菌红素(Undecylprodigiosin)合成途径中的redN基因和放线菌紫素(Actinorhodin)合成途径中的actVAORF5基因做敲除实验,结果发现pCRISPomyces-1质粒的敲除效率只有25%左右,而pCRISPomyces-2质粒无论对单基因敲除还是对多个单基因同时敲除都能达到100%的效率。另外,Cobb等还使用pCRISPomyces-2对绿色产色链霉菌的phpD和phpM基因以及白色链霉菌的sshg_05713和sshg_00040/sshg_00050进行敲除,效率达66%-100%。这也侧面说明pCRISPomyces-2在链霉菌中能够普遍适用。
Tong等[17]以温敏型质粒pGM1190为骨架,通过改造设计成pCRISPR-Cas9质粒,pCRISPR-Cas9中以tipA启动子驱动cas9/dcas9表达,PermE*启动子驱动sgRNA转录,实现精确的基因编辑。通过靶向天蓝色链霉菌(Streptomyces coelicolor A3(2))放线紫红素合成途径中的两个基因:actIORF1(SCO5087)和actVB(SCO5092)敲除去验证上述系统的可行性,研究结果发现,当没有同源修复模板时,CRISPR/Cas9系统完成对DNA的双链切割后,通过机体内NHEJ机制进行修复,在靶基因中产生一系列不同的突变体,删除效率为3%-54%,相比真核生物偏低[18]。ligase D(LigD)是NHEJ途径中的一个核心元件,绝大多数链霉菌中缺少该元件,作者将来源于肉色链霉菌(Streptomyces carneus)的LigD整合入pCRISPR-Cas9体系,显著提高了删除效率(77%);而提供同源修复模板,CRISPR/Cas9系统完成对DNA的双链切割后,机体通过HDR机制进行修复可以实现100%的精确删除。
Huang等[14]以原始质粒pKC1139为骨架设计含CRISPR/Cas9系统的质粒,其中,cas9基因由启动子tipA负责驱动,sgRNA的转录是由合成启动子j23119驱动。将其转入天蓝色链霉菌(Streptomyces coelicolorM145)中进行基因敲除,单个基因(actII-orf4、redD、glnR)敲除效率达到60%-100%。有意思的是,根据靶基因设计的不同sgRNA和培养基成分的变化对敲除效率都有不同程度的影响。同时敲除多个单基因(actII-orf4和redD)时,效率有所下降,但也达到54%。
目前在链霉菌中常使用内切核酸酶I-SceI对大片段基因进行敲除,但该方法步骤十分繁琐。CRISPR/Cas9系统能否也实现同样功能,且效率如何,Huang等给出了答案,针对放线紫红素、十一烷基灵菌红素、钙依赖抗生素合成途径的基因簇(Sco5071-5092、Sco5877-5898、Sco3210-3250) 敲除效率达54%-100%,且对多个基因簇共同敲除效率也达45%±8%。
CodA(sm)是胞嘧啶脱氨酶的D314A突变体,可以将培养基中添加的无毒性5-氟胞嘧啶(5-fluorocytosine,5FC)转化成有毒5-氟尿嘧啶(5-fluorouracil,5FU),可以被用来筛选已突变而且质粒被清除的个体。在2009年,Dubeau等[19]首次将CodA(sm)作为链霉菌的一个反选择标记。2015年,Hu等[20]将CRISPR/Cas9系统与CodA(sm)结合应用于链霉菌。他们以Streptomyces coelicolorM145中放线菌素合成途径中actI-ORF2基因为敲除对象,构建了6种不同的质粒,使用强启动子ermE*驱动sgRNA转录,同源修复模板为靶基因的上下游基因(UHA+DHA),其中不含sgRNA的质粒pWHU2656只通过同源交换,靶基因的敲除率为4%。质粒pWHU2657(scas9,sgRNA2(+48),UHA+DHA)对靶基因敲除率为93%,比pWHU2658(scas9,sgRNA2(+85),UHA+DHA)敲除率99%略低一些。将codA(sm)基因插入pWHU2659,随后把40个有安普霉素抗性重组子转到含有800 μg/mL 5FC的SFM培养基中,在135个存活子中有127个是重组子且无质粒,这大大缩减了后期筛选无质粒的突变子的周期。
雍德祥[21]通过对维吉尼亚链霉菌IBL14(Streptomyces virginiaeIBL14)的全基因组序列进行比对,发现并确定了其中的CRISPR系统属于I-B型并将其命名为CRISPR I-SV14B系统,选取设计一段引导DNA以及同源臂整合到质粒pKC1139上,通过原生质体转化的方法将其转化到IBL14中进行打靶,实验结果表明svu016基因成功地被敲除掉,且效率为70%以上。
2.1.2 CRISPRi在链霉菌中的验证 CRISPR干扰(CRISPRi)是一项基于传统CRISPR/Cas9基因编辑系统的新型基因调控工具,在某些情况下,可逆抑制基因表达策略优于基因缺失[17]。Tong等[17]以actIORF1基因为验证对象,利用CRISPRy软件对actIORF1基因的编码区设计6种sgRNA和利用sgRNAcas9软件对actIORF1启动子区域设计6个sgRNA,其中一半靶向模板链DNA,另外一半靶向非模板链(图4),研究结果发现无论当sgRNA靶向启动子区域模板链或非模板链,放线菌素合成显著降低甚至丧失。靶向ORF区域的重组子中,仅当sgRNA靶向非模板链时才观察到放线菌素量降低或者丧失。作者为了验证实验是否可逆,将重组子在37℃下培养1 d后转入新的培养基中进行孵育,结果发现重组子重新获得放线菌素合成能力。证明了CRISPRi在链霉菌中应用的可行性。

图4 用于CRISPR干扰的12个sgRNA的位置
2.2 CRISPR/Cas9系统在链霉菌中的应用
2.2.1 基因定点突变 定点突变通常包括碱基的添加、删除、点突变,它不仅可以用来阐明基因的调控机理,也可以用来研究蛋白质结构与功能之间的关系。目前常用的技术包括:寡核苷酸式诱变、寡核苷酸引物介导、重叠延伸介导和大引物诱变法介导(PCR介导)等。但这些方法均存在很明显的缺点。例如,成本高、突变效率低及操作复杂等。CRISPR/Cas9基因编辑系统因其操作方便、突变效率高等优点为基因定点突变提供了新的契机。
核糖体蛋白S12(由rpsL基因编码)的位点特异性突变使得天蓝色链霉菌对链霉素具有高的抗性(100 μg/mL)[22]。Huang[14]通过在同源修复模板上将原rpsL基因中262-264位的AAG改为GAA,利用CRISPR/Cas9系统介导成功实现基因点突变,突变效率达64%±12.7%,后期在含有100 μg/mL链霉素的平板中发现有菌落产生。
2.2.2 验证基因功能Streptomyces formicae是分离于非洲彭日格细长蚁(Tetraponera penzigiplant-ants)体内的一种新的链霉菌种,其能产生对耐甲氧西林金黄色葡萄球菌(Methicillin resistantStaphylococcus aureus,MRSA)和万古霉素耐药肠球菌(Vancomycin resistantenterococci,VRE)有活性的新型五环聚酮化合物。Qin等[23]通过对S. formicae产物鉴定分析发现,16种新的化合物分子,这些化合物具有一个核心结构Formicamycins。接着利用antiSMASH 3.0对S. formicae基因组进行分析,发现其仅存在一个二型聚酮合酶基因簇(BGC30)。随后将pCRISPomyces-2质粒转入S. formicae中,在CRISPR/Cas9系统介导下将整个BGC30敲除,后期利用LC-MS(UV)检测突变株均无Formicamycins产生。为了确定Formicamycins合成途径丢失是由于基因编辑造成,而不是由其他突变事件造成,Qin等通过对突变株回补含BGC30质粒,利用LCMS(UV)检测突变株发现突变株重新产生Formicamycins,至此,他们利用CRISPR/Cas9编辑工具确认Formicamycins是由二型聚酮合酶基因簇编码产生。
Formicamycin结构中含有4个卤素原子,在BGC30中仅存在一个单基因(forV)疑似编码卤化酶,另外在S. formicae基因组中还存在2个疑似编码卤化酶。为了鉴定ForV的生物功能,Qin等[23]利用CRISPR/Cas9系统将其敲掉,LC-MS(UV)检测发现无Formicamycin产生,从而验证了forV负责Formicamycin结构中卤素原子的引进。
2.2.3 激活链霉菌中隐性次级代谢产物生物合成基因簇 作为一类常见的细菌,链霉菌被用来产生很多作为抗生素、抗癌试剂和其他药物的化合物。然而近年来对抗生素的滥用,细菌耐药性日益严重,使得临床可用的有效抗生素越来越少。链霉菌的全基因组测序显示链霉菌中存在许多隐性次级代谢产物生物合成基因簇,在实验室条件下,它们多数处于沉默状态或者很低的表达水平,通过激活CSMG获得新抗生素已成为一个重要的研究方向。目前激活链霉菌CSMG的方法主要有:(1)改变发酵条件[24-25];(2)核糖体工程及相关策略[26];(3)联合培养[27-28];(4)异源表达[29-30];(5)遗传改造CSR(Cluster-situated regulator)基因[31-32]。在一项新的研究中,来自美国伊利诺伊大学和新加坡科技研究局的研究人员利用CRISPR-Cas9技术激活链霉菌中不表达的或者说沉默的基因簇[33]。
玫瑰孢链霉菌(Streptomyces roseosporusNRRL15998)的基因组经比对分析,其包含29个生物合成基因簇(Biosynthetic gene clusters,BGCs)[34],其中一个BGC与灰色链霉菌的PTM(Polycyclic tetramate macrolactam)基因簇序列相似性大于90%,Zhang等[33]利用CRISPR/Cas9系统介导将kasO*p插在第一个ORF的上游,成功地获得了一种新的化合物PTM2。FR-900098是只能由微红鼠灰链霉菌(Streptomyces rubellomurinus)和淡紫链霉菌(Streptomyces lavendulae)产生的一种抗疟药,Zhang等利用CRISPR/Cas9系统介导将双向P8-kasO*p启动子盒引入玫瑰孢链霉菌膦酸盐生物合成基因簇中,促使frbD操纵子和frbC同源物表达,在发酵产物中成功得到FR-900098,只是浓度低于微红鼠灰链霉菌的发酵液。随后,他们利用同样的方法在绿色产色链霉菌中将kasO*p插入到一个II型PKS基因簇的最前端,成功激活BGC表达,产生一种新的化合物 4(C23H16O8)。
3 CRISPR/Cas9系统在链霉菌应用的潜力
晶体学技术为解析蛋白结构和分子作用机制提供很好的手段,从而帮助人们对Cas9酶进行优化,使其可以完美发挥基因编辑效果。CRISPR/Cas9系统在行使基因编辑功能时,cas9中C端结构域(PAM-interacting,PI)起着特异性识别作用,不同的PI识别不同的PAM区,具有专一性,但这限制了其可靶向序列的范围。目前应用最为普遍的化脓性链球菌Cas9(SpCas9)和金黄色葡萄球菌Cas9(SaCas9),其识别的PAM序列分别为 5'-NGG-3'和5'-NNGRRT-3'。基于PI对PAM的特异性具有决定作用,在2015年,Kleinstiver[35-36]研究小组分别对SpCas9和SaCas9进行改造,共得到3种SpCas9突变体和1种SaCas9突变体,它们可以识别更广泛的核苷酸序列,靶向以往CRISPR-Cas9技术无法触及的基因组位点,同样,其他一些研究小组也获得了成功[37](表2)。此项研究扩大了CRISPR-Cas9工具箱的靶向空间,为CRISPR-Cas9系统在链霉菌或其他物种中应用提供了更多的可能性。

表2 几种不同cas9的信息
4 问题与展望
CRISPR/Cas9系统虽然能够高效快速地进行基因编辑,为基础研究提供了强大的平台。但该技术的也存在一个最大的缺点即脱靶效应,所谓“脱靶效应”[49],简单来说,就是编辑了“预想之外”的基因,带来各种不可控的不良后果。最近,发表在Nature Methods杂志上题为“Unexpected mutations after CRISPR-Cas9 editingin vivo”的论文称,CRISPR能够引入数百种意想不到的突变到基因组中[50],但由于其样本量选择问题受到很多专家质疑。值得庆幸的是,最近的研究表明CRISPR/Cas9系统在细菌中是特异的,而且在链霉菌中目前也未有报道有关脱靶问题。在其他物种中针对脱靶问题,近年来科学家们研发了不少检测脱靶剪切酶活性的实验方法,比如 GUIDE-seq[51]、Digenome-seq[52]、基于整合酶缺陷型慢病毒载体(IDLV)的技术[53]、BLESS[54]、SITE-Seq[55]、CIRCLE-seq[56],这些方法各有优劣势,但都可以很好地预测潜在的脱靶位点。
链霉菌能够产生多种抗生素,而且多数为非致病菌,所以长期以来都是生命科学与医学、药学以及工业领域的研究热点,然而面对抗生素“瓶颈时代”的到来,这就需要我们在原有研究基础上重新开启抗生素研究的新路径、新方法、新思维。近年来随着分子生物学和基因组测序技术的不断发展以及合成生物学概念的提出[57],使人们能够利用已有的基因组信息和分子生物学方法明确地对目标菌株进行改造。基于CRISPR/Cas9基因编辑系统,我们可以从两方面着手,一是可以利用该技术干扰链霉菌中其他代谢旁路来提高目的产物的产量和质量;二是可以利用该技术实现基因敲除或者对链霉菌中PKS或NRPS局部替换从而获得新的代谢产物从而达到微生物定向育种的目的。本课题组目前正在利用CRISPR/Cas9系统对产子囊霉素的吸水链霉菌中的PKS或NRPS局部模块替换,希望得到产他克莫司的突变菌株。CRIPSR除了用于精准编辑某个基因,还能用于调控基因的表达,包括CRISPRi和CRISPR激活(CRISPRa)。cDNA过表达是上调个别基因的表达水平的常用方法,与这种方法相比,CRISPRa使用起来更加简单,而且可以同时激活多个基因,MIT的CRISPR先驱张锋通过改造dCas9和sgRNA优化了转录机器的招募,成功将RNA表达水平提升了一个数量级。链霉菌的染色体基因组12%为调控基因[58],其中比较典型的包括链霉菌抗生素调节蛋白(Streptomycesantibiotic regulatory protein,SARP)家族和LuxR家族等,大多数调控基因与链霉菌的初级及次级代谢有关,过表达链霉菌中正调控基因是提高抗生素产量的常用策略,基于CRISPR/Cas9系统的简便性,科学家们已利用CRISPRa技术提高细胞中正调控基因的转录水平,遗憾的是,CRISPRa技术还未在链霉菌中得到应用。随着CRISPR/Cas9技术的不断优化,相信在不久的将来,通过CRISPR/Cas9介导在链霉菌中获得新抗生素将会迎来新的契机。
[1]Lucas X, Senger C, Erxleben A, et al. StreptomeDB:a resource for natural compounds isolated fromStreptomycesspecies[J]. Nucleic Acids Res, 2013, 41(Database issue):1130-1136.
[2]Campo N, Daveranmingot ML, Leenhouts K, et al.Cre-loxP recombination system for large genome rearrangements inLactococcus lactis[J]. Appl Environ Microbiol, 2002, 68(5):2359-2367.
[3]Oliynyk M, Stark CB, Bhatt A, et al. Analysis of the biosynthetic gene cluster for the polyether antibiotic monensin inStreptomyces cinnamonensisand evidence for the role ofmonBandmonCgenes in oxidative cyclization[J]. Mol Microbiol, 2003, 5:1179-1190.
[4]Zhou M, Jing X, Xie P, et al. Sequential deletion of all the polyketide synthase and nonribosomal peptide synthetase biosynthetic gene clusters and a 900-kb subtelomeric sequence of the linear chromosome ofStreptomyces coelicolor[J]. FEMS Microbiol Lett,2012, 333(2):169-179.
[5]Gust B, Challis GL, Fowler K, et al. PCR-TargetedStreptomycesgene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin[J]. Proc Natl Acad Sci USA,2003, 100(4):1541-1546.
[6]Sternberg N, Hamilton D. Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites[J]. J Mol Biol, 1981, 150(4):467-486.
[7]Volkert FC, Wilson DW, Broach JR. Deoxyribonucleic acid plasmids in yeasts[J]. Microbiol Rev, 1989, 53(3):299-317.
[8]Sauer B, Mcdermott J. DNA recombination with a heterospecific Cre homolog identified from comparison of the pac-c1 regions of P1-related phages[J]. Nucleic Acids Res, 2004, 20:6086-6095.
[9]Raynal A, Karray F, Tuphile K, et al. Excisable cassettes:New tools for functional analysis ofStreptomycesgenomes[J]. Appl Environ Microbiol, 2006, 72(7):4839-4844.
[10]Le C, Ran FA, Cox D, et al.Multiplex genome engineering using CRISPR/Cas systems[J]. Science, 2013, 6121 :819-823.
[11]Cheng AW, Wang H, Yang H, et al.Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system[J]. Cell Res, 2013, 23(10):1163-1171.
[12]Siegl T, Luzhetskyy A. Actinomycetes genome engineering approaches[J]. Antonie Van Leeuwenhoek, 2012, 102(3):503-516.
[13]Fedoryshyn M, et al.Marker removal from actinomycetes genome using Flp recombinase[J]. Gene, 2008, 419(1-2):43-47.
[14]Huang H, Zheng G, Jiang W, et al. One-step high-efficiency CRISPR/Cas9-mediated genome editing inStreptomyces[J].Acta Biochimica et Biophysica Sinica, 2015, 47(4):231-243.
[15]Cobb RE, Wang Y, Zhao H. High-efficiency multiplex genome editing ofStreptomycesspecies using an engineered CRISPR/Cas system[J]. Acs Synthetic Biology, 2015, 4(6):723-728.
[16]Leskiw BK, et al. The use of a rare codon specifically during development?[J]. Mol Microbiol, 1991, 12 :2861-2867.
[17]Tong Y, Charusanti P, Zhang L, et al. CRISPR-Cas9 Based engineering of actinomycetal genomes[J]. Acs Synthetic Biology,2015, 4(9):1020-1029.
[18]Bowater R, Doherty AJ. Making ends meet:repairing breaks in bacterial DNA by non-homologous end-joining[J]. PLoS Genet,2006, 2(2):e8.
[19]Dubeau MP, Ghinet MG, Jacques PE, et al. Cytosine deaminase as a negative selection marker for gene disruption and replacement in the genusStreptomycesand other actinobacteria[J]. Appl Environ Microbiol, 2009, 75(4):1211-1214.
[20]Hu Z, Shishi W, Wei X, et al. Highly efficient editing of the actinorhodin polyketide chain length factor gene inStreptomyces coelicolorM145 using CRISPR/Cas9-CodA(sm)combined system[J]. Appl Microbiol Biotechnol, 2015, 24:10575-10585.
[21]雍德祥. 维吉尼亚链霉菌IBL14中的CRISPR-Cas9系统及其基因编辑方法[D]. 合肥:安徽大学, 2016.
[22]Shima J, et al.Induction of actinorhodin production by rpsL(encoding ribosomal protein S12)mutations that confer streptomycin resistance inStreptomyceslividansandStreptomyces coelicolor A3(2)[J]. J Bacteriol, 1996, 178(24):7276-7284.
[23]Qin Z, et al.Formicamycins, antibacterial polyketides produced byStreptomyces formicaeisolated from AfricanTetraponeraplantants[J]. Chem Sci, 2017, 8(4):3218-3227.
[24]Scherlach K, Hertweck C. Discovery of aspoquinolones A-D,prenylated quinoline-2-one alkaloids fromAspergillus nidulans,motivated by genome mining[J]. Org Biomol Chem, 2006, 4(18):3517-3520.
[25]Craney A, Ozimok C, et al.Chemical perturbation of secondary metabolism demonstrates important links to primary metabolism[J]. Chem Biol, 2012, 19(8):1020-1027.
[26]Imai Y, Sato S, et al. Lincomycin at subinhibitory concentrations potentiates secondary metabolite production byStreptomycesspp[J]. Appl Environ Microbiol, 2015, 11:3869-3879.
[27]Onaka H, Mori Y, Igarashi Y, et al.Mycolic acid-containing bacteria induce natural-product biosynthesis inStreptomycesspecies[J]. Appl Environ Microbiol, 2011, 77(2):400-406.
[28]Moody SC. Microbial co-culture:harnessing intermicrobial signaling for the production of novel antimicrobials[J]. Future Microbiol, 2014, 9(5):575-578.
[29]Martin R, Sterner O, et al. ChemInform abstract:collinone, a new recombinant angular polyketide antibiotic made by an engineeredStreptomycesstrain[J]. J Antibiotics, 2001, 54(3):239-249.
[30]Gomezescribano JP, Bibb MJ. EngineeringStreptomyces coelicolorfor heterologous expression of secondary metabolite gene clusters[J]. Microbial Biotechnology, 2011, 4(2):207-215.
[31]Laureti L, Aigle, Khosla C. Identification of a bioactive 51-membered macrolide complex by activation of a silent polyketide synthase inStreptomycesambofaciens[J]. Proc Natl Acad Sci USA, 2011, 108(15):6258-6263.
[32]Zhou Z, Xu Q, Bu Q, et al. Genome mining-directed activation of a silent angucycline biosynthetic gene cluster inStreptomyces chattanoogensis[J]. Chembiochem, 2015, 16(3):496-502.
[33]Zhang MM, Wong FT, Wang Y, et al. CRISPR-Cas9 strategy for activation of silentStreptomycesbiosynthetic gene clusters[J].Nat Chem Biol, 2017. doi:10.1038/nchembio.2341.
[34]Weber, Tilmann, Charusanti, et al. Metabolic engineering of antibiotic factories:new tools for antibiotic production in actinomycetes[J]. Trends Biotechnol, 2015, 33(1):15-26.
[35]Kleinstiver BP, Prew MS, Tsai SQ, et al.Engineered CRISPR-Cas9 nucleases with altered PAM specificities[J]. Nature, 2015, 523(7561):481-485.
[36]Kleinstiver BP, Prew MS, Tsai SQ, et al. Broadening the targeting range ofStaphylococcus aureusCRISPR-Cas9 by modifying PAM recognition[J]. Nat Biotechnol, 2015, 33(12):1293-1298.
[37]Hirano S, Nishimasu H, Ishitani R, et al. Structural basis for the altered PAM specificities of engineered CRISPR-Cas9[J]. Mol Cell, 2016, 61(6):886-894.
[38]Hirano H, Gootenberg JS, Horii T, et al.Structure and engineering ofFrancisella novicidaCas9[J]. Cell, 2016, 5:950-961.
[39]Mojica FJ, Díez-Villaseñor C, García-Martínez J, et al. Short motif sequences determine the targets of the prokaryotic CRISPR defence system[J]. Microbiology, 2009, 155(Pt 3):733-740.
[40]Jinek M, Jiang F, Taylor DW, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation[J].Science, 2014, 343(6176):1247997.
[41]Jiang F, Zhou K, Ma L, et al. STRUCTURAL BIOLOGY. A Cas9-guide RNA complex preorganized for target DNA recognition[J].Science, 2015, 348(6242):1477-1481.
[42]Anders C, Bargsten K, Jinek M. Structural plasticity of PAM recognition by engineered variants of the RNA-guided endonuclease Cas9[J]. Mol Cell, 2016, 61(6):895-902.
[43]Horvath P, Romero DA, Coute-Monvoisin AC, et al.Diversity,activity, and evolution of CRISPR loci inStreptococcus thermophilus[J]. J Bacteriol, 2008, 190(4):1401-1412.
[44]Fonfara I, Le RA, Chylinski K, et al. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems[J]. Nucleic Acids Res,2014, 42(4):2577-2590.
[45]Ran FA, et al.In vivogenome editing usingStaphylococcus aureusCas9[J]. Nature, 2015, 520(7546):186-191.
[46]Nishimasu H, Cong L, Yan WX, et al. Crystal Structure ofStaphylococcus aureusCas9[J]. Cell, 2015, 5:1113-1126.
[47]Zhang Y, Heidrich N, Ampattu BJ, et al. Processing-independent CRISPR RNAs limit natural transformation inNeisseria meningitidis[J]. Mol Cell, 2013, 50(4):488-503.
[48]Yamada M, et al. Crystal structure of the minimal Cas9 fromCampylobacter jejunireveals the molecular diversity in the CRISPRCas9 systems[J]. Mol Cell, 2017, 65(6):1109-1121.
[49]Ramalingam S, Annaluru N, Chandrasegaran S. A CRISPR way to engineer the human genome[J]. Genome Biol, 2013, 14(2):1-4.
[50]Schaefer KA, et al.Unexpected mutations after CRISPR-Cas9 editingin vivo[J]. Nat Methods, 2017, 14(6):547-548.
[51]Zhu LJ, Lawrence M, Gupta A, et al.GUIDEseq:a bioconductor package to analyze GUIDE-Seq datasets for CRISPR-Cas nucleases[J]. BMC Genomics, 2017, 18(1):379-388.
[52]Park J, Childs L, Kim D, et al.Digenome-seq web tool for profiling CRISPR specificity[J]. Nat Methods, 2017, 14(6):548-549.
[53]Wang X, Wang Y, Wu X, et al. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors[J]. Nat Biotechnol, 2015, 33(2):175-178.
[54]Nicola C, Abhishek M, Joao SM, et al. Nucleotide-resolution DNA double-strand breaks mapping by next-generation sequencing[J].Nat Methods, 2013, 10(4):361-365.
[55]Cameron P, Fuller CK, Donohoue PD, et al. Mapping the genomic landscape of CRISPR-Cas9 cleavage[J]. Nat Methods, 2017, 14(6):600-606.
[56]Tsai SQ, Nguyen NT, Malagon-Lopez J, et al. CIRCLE-seq:a highly sensitivein vitroscreen for genome-wide CRISPR-Cas9 nuclease off-targets[J]. Nat Methods, 2017, 14(6):607-614.
[57]Heinemann M, Panke S. Synthetic biology--putting engineering into biology[J]. Bioinformatics, 2006, 22(22):2790-2799.
[58]Romero-Rodríguez A, Robledo-Casados I, Sánchez S. An overview on transcriptional regulators inStreptomyces[J]. Biochim Biophys Acta, 2015, 1849(8):1017-1039.
猜你喜欢
中国计量大学学报(2022年2期)2022-07-18
当代水产(2022年1期)2022-04-26
西北农林科技大学学报(自然科学版)(2019年8期)2019-07-17
食品科学(2018年10期)2018-05-23
西南医科大学学报(2015年1期)2015-08-22
少儿科学周刊·少年版(2015年3期)2015-07-07
少儿科学周刊·少年版(2015年3期)2015-07-07
中国当代医药(2015年9期)2015-03-01
遗传(2015年5期)2015-02-04
西南军医(2015年6期)2015-01-23
