
SIRT1与类风湿关节炎的研究进展
2019-10-16 06:50综述审校
安徽医科大学学报 2019年9期
柳 晓 综述 徐 涛 审校
类风湿关节炎(Rheumatoid arthritis,RA)是一种慢性全身性自身免疫性疾病,以对称性、进行性多关节滑膜炎为主要特征,已经成为一种世界性疾病。RA发病高峰在20~45岁,女性多发,我国的患病率约为0.003 5~0.004 5[1]。复杂的病理过程不仅减少患者的寿命,同时也对患者自身经济情况造成负担。RA的主要病理改变是关节滑膜的慢性炎症和增生,从而导致血管翳的形成,侵蚀关节软骨、软骨下骨、韧带等,破坏关节软骨、骨和关节囊,最终致使关节畸形和功能障碍。其过程中伴有多种关节外表现,如发热、皮下结节和淋巴结肿大等,血清中也可出现多种自身抗体,病变呈持续、反复发作过程[2]。免疫因素、感染因素、遗传因素以及环境因素等都是引起该病发生的关键因素。最近,有研究[3-4]已经检测到RA患者中沉默信息调节因子1(silence information regulator 1,SIRT1)的异常表达。其中有研究[5]发现,SIRT1在RA患者成纤维样滑膜细胞(fibroblast-like synovial cells,FLS)和滑膜组织中均高表达。FLS中SIRT1的过表达可以促进炎症因子的释放以及抑制FLS的凋亡。而另一方面,沉默FLS中SIRT1的表达,可以减轻FLS诱发的炎症反应以及增加FLS凋亡,充分说明SIRT1对RA存在影响和在RA治疗中的潜力。本文总结了SIRT1在RA发生发展中对免疫细胞、滑膜细胞、骨组织等方面的具体作用机制,并对SIRT1作为治疗靶点的潜在可行性进行探讨。
1 Sirtuin家族和SIRT1
SIRT1属于Sirtuin蛋白家族,存在于多种生物中。根据X线晶体衍射显示,Sirtuin蛋白具有高度的保守性。第一种被发现的Sirtuin蛋白家族成员是沉默信息调节因子2蛋白(Sir2),由于它存在于酿酒酵母细胞内,Sirtuin也因此得名。哺乳动物中已经有7种Sirtuin蛋白被鉴定出来。它们具有不同的定位和功能,SIRT1、SIRT6和SIRT7主要分布于细胞核,而SIRT2主要存在于细胞质,其他三种SIRT3、SIRT4和SIRT5蛋白分布于线粒体。其中有表现出ADP核糖转移酶活性,也有表现出NAD+依赖的去乙酰化酶活性,或二者兼有。在低等生物体中,Sirtuin蛋白具有调节寿命的作用。而在高等生物中,其参与的生命活动非常多样:细胞应激反应,DNA损伤修复、调节基因表达、代谢和存活等,Sirtuin还被发现具有复杂的免疫调节作用,在自身免疫疾病中发挥重要作用[6]。
SIRT1是Sir2的哺乳动物同源体,属于Ⅲ类组蛋白去乙酰化酶。它定位于细胞核中,但是可作用于细胞质靶点。SIRT1和其他Sirtuin蛋白类似,其核心催化区同样由275个氨基酸组成。此外,构成人体SIRT1蛋白的500个氨基酸残基中,去乙酰化活性的必需基团是第363位组氨酸。SIRT1可以去乙酰化组蛋白H1第26位、H3第9位、H4第16位赖氨酸。同时,SIRT1也可通过去乙酰化调节多种转录因子活性,如核蛋白SIRT1作用于FOXO1、FOXO3、PGC-1α、p53等。而在细胞质主要调节核转录因子-κB (nuclear factor-κB,NF-κB)、HIF1α、Notch等。正是通过这些作用,SIRT1具有调节DNA修复、细胞代谢、自噬、细胞周期和炎症等多种生理功能。

表1 SIRT1的介绍及其在RA中的功能
2 SIRT1对免疫细胞的调节作用
由于RA的发病机制过于复杂,其理论一直处于发展中,因此确切病因及发病机制尚未明确。但随着对RA更深入的研究发现,在RA患者的滑膜组织和滑膜液中,出现巨噬细胞、T细胞等细胞免疫调节的异常,故目前多认为其是导致发病的关键因素。然而,近期文献[7]报道SIRT1作用于免疫系统。胸腺、脾脏和淋巴结以及纯化的辅助性T细胞(CD4+T细胞)中均能检测到SIRT1出现高表达的现象,表明SIRT1影响T细胞增殖分化从而参与免疫反应。
2.1 SIRT1与巨噬细胞巨噬细胞是固有免疫系统中最主要的免疫细胞,由外周血单核细胞分化而来,二者在RA的发病过程中起着关键性的作用。在炎症反应中,巨噬细胞主要作用是在RA滑膜里局部增生,被活化T细胞分泌的细胞因子激活,释放大量促炎性细胞因子肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)、白介素6(interleukin-6,IL-6)、白介素1(interleukin-1,IL-1)等。其中TNF-α是目前研究最多且最为深入的细胞因子,也是RA发病机制中关键的促炎因子。TNF-α刺激软骨细胞合成IL-6与白介素8(interleukin-8,IL-8),进一步引起炎性反应的发生,以致血管翳的形成及软骨坏死。而血管翳是RA病变过程中的重要病理产物,并且是导致关节病变、骨和软骨破坏的主要原因及病理基础。此外,活化的巨噬细胞引起组织相容性复合体Ⅱ分子过度表达,分泌大量的细胞黏附分子、趋化因子、细胞因子、生长因子和蛋白酶等,而这些炎症介质共同参与到RA病理机制。
在ApoE 基因缺陷小鼠实验中,用SIRT1激动剂干预其巨噬细胞,发现细胞间黏附分子及炎症因子表达水平降低。在胶原诱导的大鼠关节炎模型中,SIRT1通过去乙酰化 p65 和 p300 降低NF-κB转录活性,抑制由NF-κB信号通路活化介导的炎症因子的合成与分泌,减轻关节炎症。验证了SIRT1可通过抑制NF-κB信号通路来减轻巨噬细胞的促炎症表型。不仅如此,SIRT1通过抑制激活蛋白-1(activator protein-1,AP-1)的转录活性和下游基因COX-2的表达,来调控由巨噬细胞介导的炎症反应。此外,在腹膜巨噬细胞中,过表达的SIRT1可以降低COX-2 mRNA 的表达水平,进而下调巨噬细胞分泌前列腺素E(PGE),以此来增强抗炎以及对肿瘤的杀伤作用。并且,SIRT1抑制巨噬细胞中未折叠蛋白的产生,从而调控巨噬细胞活性。除此之外,Hah et al[8]发现SIRT1缺失的巨噬细胞增加了M1极化,增加了促炎细胞因子的产生,SIRT1转基因小鼠巨噬细胞表现为M2表型巨噬细胞极化的增强和M1表型巨噬细胞极化的减弱,并且伴随抗炎因子的增多与促炎因子的减少。因此,SIRT1可以通过调节M1/M2极化,从而抑制RA炎症反应,促进组织修复。
2.2 SIRT1与T细胞T细胞是RA滑膜组织中的主要炎性细胞。在RA的自身免疫反应中,其机制主要是自身抗原被MHC Ⅱ型阳性的抗原呈递细胞(APCs)传递给CD4+T细胞,从而启动免疫性应答。CD4+T细胞在不同的特异性转录因子调控下,分化为不同的细胞亚群,包括Th1、Th2、Th17和调节性T细胞(Treg)等细胞。Th1细胞主要介导细胞免疫,在APCs活化后,主要分泌IFN-γ、IL-2、TNF-α和TNF-β。区别于Th1细胞,Th2细胞主要介导体液免疫,Th2和Treg细胞主要分泌抗炎细胞因子,并且Treg细胞在介导免疫稳态、维持免疫耐受及控制疾病进展方面起重要作用。而相关的Th1细胞因子可以抑制初始T细胞分化为Th2细胞并同时抑制产生相关抗炎细胞因子,以至抗炎细胞因子相对缺乏而前炎症因子过量,最终造成Th1/Th2细胞失衡。RA的发生被认为与这种失衡密切相关[9]。此外,分泌高水平IL-17的Th17细胞具有致炎、促血管新生导致血管翳形成、破坏骨和软骨作用,从而导致RA的发生发展[10]。
体外实验和动物实验均显示,在SIRT1被沉默之后,T细胞更容易被激活、分化成效应T细胞。而多种AP-1和NF-κB靶基因也因过度活化的T细胞对AP-1和NF-κB转录的抑制作用减弱而均能检测到上调。其中SIRT1主要是通过去乙酰化c-Jun来抑制AP-1信号通路,从而抑制T细胞活化和增殖,以及IL-2的产生。除了直接调节AP-1、NF-κB等转录因子的活性之外,SIRT1也可以调节影响T 细胞增殖和功能的相关基因。如 SIRT1可以抑制T细胞活化所必须一种结合蛋白Bclf1的转录,控制过度增殖和异常活化的T细胞。同时,SIRT1可以抑制STAT3的激活从而调节CD4+T细胞分化。T细胞被IL-6、IL-21等细胞因子刺激后,STAT3也随之被JAK-STAT信号通路激活并进行转录,从而调节CD4+T细胞向Th2或Th17细胞方向分化。此外,STAT3激活后,还能通过抑制小鼠叉头框蛋白P3(FXOP3)来抑制CD4+T细胞的表达。并且IL-6刺激STAT3后可以上调趋化因子(如CCL5),促进T细胞迁移到炎症部位,表明STAT3与促炎症T细胞表型相关。即SIRT1通过抑制STAT3调节CD4+T细胞分化。对于FXOP3,有研究[11]发现SIRT1能直接去乙酰化FXOP3或者通过去乙酰p65而促进FXOP3的去乙酰化,其结果是SIRT1通过降低p65和FXOP3乙酰化,减少FXOP3表达并降低其稳定性,进而抑制T淋巴细胞分化。此外,相比活化的T细胞或幼稚的T细胞,SIRT1 mRNA及其蛋白在无反应T细胞中的表达水平明显升高,这表明SIRT1在维持T细胞耐受中具有潜在作用。因此,SIRT1可以通过多种途径调节T细胞活化和增殖以及CD4+T细胞的分化,从而抑制RA炎症反应。
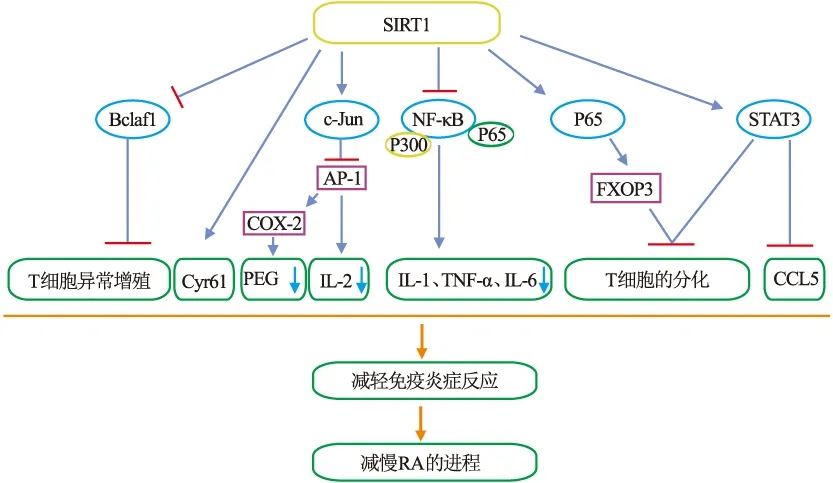
图1 SIRT1在RA中的免疫调节作用
3 SIRT1对滑膜细胞的作用
滑膜细胞增生是RA一个重要的病理特点,它与新生微血管、炎性细胞及机化的纤维素共同构成RA病程中具有侵袭性的滑膜血管翳,分泌多种蛋白酶降解周围的骨质与软骨,直接破坏软骨的结构。人类正常滑膜组织衬里层的细胞分为两种类型:巨噬细胞样滑膜细胞(A型)和成纤维细胞样滑膜细胞(B型,即FLS)。FLS是滑膜增生的主要细胞,并且具有肿瘤细胞样侵袭和迁移能力,在RA病理状态下呈现异常增殖和凋亡抑制状态。FLS不仅参与软骨的破坏,还分泌多种细胞因子、趋化因子以及血管生成因子,在RA滑膜病理过程中起着关键性作用[12-13]。因此,通过抑制滑膜细胞侵袭和迁移能力促进滑膜细胞凋亡,抑制滑膜细胞增殖与炎性因子释放,可有效控制关节变形与滑膜炎症,保护关节软骨和骨,减缓RA病程进展。
最近,已经检测到RA患者FLS和滑膜组织中SIRT1的异常表达。其中发现SIRT1的激活剂白藜芦醇(Res)能促进caspase-3活化,诱导FLS细胞凋亡。同时发现Res能降低FLS细胞中Akt磷酸化水平,抑制3-磷酸肌醇激酶/蛋白激酶B信号通路的活化,减少其下游磷酸化相关死亡促进因子Bad抗原(Bcl-xl/Bcl-2 associated death promoter,BAD)的表达,从而诱导FLS细胞发生凋亡。Engler et al[14]发现沉默SIRT1降低了FLS的增殖率与潜在的黏附性。沉默NF-κB p65基因抑制FLS的增殖,并诱导其凋亡。 而p53也被证明在RA滑膜中过表达,并有助于调节滑膜细胞的侵袭、凋亡和增殖, SIRT1可以去乙酰化NF-κB与p53并且抑制其下游基因[15],因此这提示SIRT1可通过去乙酰化NF-κB p65与p53 来促进FLS凋亡,并抑制其增殖。在胶原诱导的大鼠关节炎模型中显示,SIRT1是诱导滑膜纤维细胞中参与RA发病机制重要细胞因子人富含半胱氨酸蛋白61(cysteine-rich protein 61,Cyr61)的关键[16]。Cyr61可以直接调节细胞黏附和迁移过程,同时又可通过自分泌和旁分泌调节反馈回路的其他细胞因子和趋化因子的产生。由于RA滑膜细胞似肿瘤样增殖并异常分泌炎性因子导致软骨破环,软骨凋亡退变释放的降解片段引起滑膜细胞的炎症反应。而SIRT1通过抑制滑膜NF-κB转录,从而抑制IL-1、TNF-α、IL-6等下游因子的转录水平,打破了这种滑膜-软骨的恶性循环。除此之外,滑膜微血管增生是由于滑膜细胞缺氧诱导因子-1(HIF-1)的表达,从而促进一些炎症细胞因子、促血管生成因子和基质金属蛋白酶产生,在RA进程中发挥重要作用。上调的SIRT1通过去乙酰化HIF-1使其失活,而HIF-1与促进血管生成因子VEGF基因启动子区域的低氧反应元素结合激活了低氧细胞中VEGF的转录,因此 SIRT1-HIF-1-VEGF 信号通路有效地减少了滑膜细胞的缺氧反应,也从根本上抑制了滑膜血管因缺氧而过度增生。
4 SIRT1对骨组织的作用
RA患者晚期常常会出现多关节软骨和骨的侵蚀性破坏,其中骨密度下降直至骨质疏松是RA患者的常见临床合并症,并最终导致关节畸形和严重的功能障碍,甚至致残。而导致RA患者骨折的主要原因正是全身性骨质疏松。RA患者的骨质疏松程度较健康对照人群显著升高,并且炎症活动与骨代谢相关联RA患者骨密度明显低于健康人群。使用SIRT1杂合缺失的雌性小鼠表现出明显的骨密度下降。另一方面,Edwards et al[17]观察发现,成骨细胞特异性SIRT1基因敲除小鼠减少成骨细胞分化,导致骨量下降,同时破骨细胞特异性SIRT1基因敲除小鼠激活NF-κB信号通路以促进破骨细胞分化至成熟,从而导致骨量下降。而抑制NF-κB信号通路则可以逆转骨量的下降,说明SIRT1可以凭借抑制NF-κB信号通路维持正常骨骼的形成,同时在骨关节病的发病中发挥重要作用。NF-κB受体活化因子(RANK)/NF-κB受体活化因子配体(RANKL)系统是调节破骨细胞分化、成熟和凋亡最重要的信号通路。RANKL通过结合破骨细胞表面的RANK,进一步激活NF-κB、p38、JNK和ERK等信号通路,达到介导滑膜巨噬细胞向破骨细胞分化与成熟的目的,引起骨质丢失[18]。而STAT3可以促进IL-6家族炎症因子的释放,同时诱导RANKL的表达,最终导致关节的破坏。然而,由于SIRT1在骨细胞中发挥重要作用,并且可以下调STAT3抑制糖异生的作用。因此SIRT1可以通过下调STAT3来减缓关节的破环。
除此之外,基质金属蛋白酶13(matrix metalloproteinase 13,MMP-13)缺失的关节炎进展明显减慢,而过表达的MMP-13不仅促进炎症反应,也直接破坏关节软骨[19]。MMP是分解关节软骨胶原的重要的软骨胶原酶,而作为内源性调控性抑制剂金属蛋白酶组织抑制因子1(tissue inhibitor of metalloproteinase-1,TIMP-1)抑制胶原的分解。与之类似的是,丁永利 等[20]发现过表达的SIRT1可以下调MMP-13 mRNA 的表达,上调TIMP-1 mRNA的表达,表明SIRT1通过调节MMP及TIMP的平衡,来降低软骨受损程度和抑制软骨的退化。由于一些细胞因子也参与软骨细胞的修复与凋亡活动,如IL-1β和TNF-α 都可上调MMP基因mRNA表达,抑制软骨基质中糖蛋白和胶原合成,增强软骨基质降解,还可诱导产生一氧化氮,抑制蛋白聚糖及Ⅱ型胶原合成,诱导软骨细胞凋亡,IL-6不仅通过自分泌形式影响软骨和滑膜细胞的正常增殖,还可协同 IL-1 阻碍成纤维细胞胶原合成,抑制软骨细胞的合成修复[21]。而大量的文献[22]都表明活化的SIRT1通过去乙酰化NF-κB p65与p53,来抑制IL-1β、TNF-α、IL-6等细胞因子诱导的关节软骨细胞的炎症反应与软骨细胞的凋亡。
因此,SIRT1能通过调节骨组织中成骨细胞、破骨细胞以及软骨细胞的分化、成熟与凋亡来抑制关节软骨和骨的侵蚀性破坏,从而促进关节炎症的恢复,抑制骨密度的下降,降低RA患者致残率。
5 展望
研究发现,除RA之外,SIRT1还作用于其他免疫疾病。如系统性红斑狼疮中,SIRT1能发挥其促进或抑制的作用。国外研究[23]已经证明SIRT1可以对其他自身免疫疾病如系统性红斑狼疮中B细胞进行调节。也有研究[24]显示调节性B细胞(regulatory B cells, Bregs)可能具有免疫调节,抑制RA进程的作用。在此基础上,SIRT1和Bregs的联系以及SIRT1与Bregs能否共同对RA进程进行抑制都值得进一步去研究。这些疑问都说明RA中SIRT1的治疗潜力值得关注,并需要更多的研究来进一步说明SIRT1和RA之间的关系。
已有研究[25]表明竞争性IL-1受体拮抗剂(IL-1RA, ANAKYRA)能缓解RA引发的疼痛感。但由于其多效作用和使用剂量的不规范性,这些治疗可能具有重大的副作用。最近Brunger et al[26]使用CRISPR/CAS9技术,对促炎性趋化因子Ccl2的起始密码子基因位点进行靶向性IL-1和TNF-α拮抗剂的基因添加,构建具有指定的炎性细胞因子抗性特征的多能干细胞,其以自动调节、反馈控制的方式拮抗IL-1或TNF-α介导的炎症,从而减轻关节炎症。由于SIRT1在RA发展中发挥重要作用,因此通过探索SIRT1的转基因位点,并将SIRT1靶向性地添加,定制干细胞内的固有细胞信号通路,为RA提供更安全和更有效的创新性治疗途径。
此外,近年来研究[27]显示,RA的发病与患者肠道菌群生态失调有着密切联系。并且发现RA患者粪便中双歧杆菌和脆弱类杆菌与正常组相比,数量明显偏低。Wellman et al[28]研究发现肠道菌群的改变是导致结肠炎症的主要原因,并且表明了SIRT1通过直接调节乳酸杆菌来调节肠道微生物群的稳态,进而改善肠道炎症和对传染性法氏囊病的易感性。因此通过SIRT1调节肠道微生物群的稳态可以为RA患者提供一种新的治疗或预防方法,但其具体机制仍需进一步研究。
6 结论
综上所述,SIRT1紧密作用于RA中,SIRT1通过负向调控多条信号通路来减缓RA的发生发展过程,主要包括调控免疫细胞与免疫因子的活化与释放,从而调控RA的免疫反应;调控骨组织中成骨细胞、破骨细胞以及软骨细胞的分化成熟与凋亡;抑制FLS的增生,减少血管翳的生成,从而减少致畸率。若SIRT1活性下降或缺失,则可以增加炎症与自身免疫性疾病的发病概率。这些研究结果提示SIRT1可以作为治疗RA的潜在靶点。
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08
材料与冶金学报(2022年2期)2022-08-10
舰船科学技术(2022年10期)2022-06-17
建材发展导向(2021年14期)2021-08-23
云南化工(2020年11期)2021-01-14
作文成功之路·小学版(2020年6期)2020-07-27
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
中国组织化学与细胞化学杂志(2016年3期)2016-02-27
中国当代医药(2015年16期)2015-03-01
西南医科大学学报(2014年6期)2014-03-20
