
苯丙素苷Acteoside,Isoacteoside和Ligupurpuroside J的全合成
2020-08-26 03:11胡志飞魏炳成
高等学校化学学报 2020年8期
胡志飞,徐 鹏,魏炳成,俞 飚
(1.上海科技大学物质科学与技术学院,上海201210;2.中国科学院上海有机化学研究所生命有机化学国家重点实验室,上海200032)
苯丙素苷(PPGs)是一类含有C6—C3芳香核片段的天然糖苷类化合物,广泛存在于植物中.对该类糖苷的研究始于上世纪60年代,目前已有相当数量并具有各类生理活性的苯丙素苷被分离鉴定[1~8].苯丙素苷的核心糖单元是β-D-吡喃葡萄糖,其糖环上不同位置羟基可以被不同基团修饰:异头位羟基常连接取代苯乙基作为苷元;2-,4-或6-位羟基可连接反式取代肉桂酰基;糖环的2-,3-,4-或6-位羟基常连接有L-鼠李糖(Rha)、D-半乳糖(Gal)、D-木糖(Xyl)和D-葡萄糖(Glc)等单糖或二糖,形成结构更为复杂的苯丙素苷(Scheme 1).活性研究结果表明,苯丙素苷具有抗菌消炎[9]、抗肿瘤[10~12]、抗病毒[13]、抗氧化[14~16]及保肝护肝[17,18]等生物活性.由于该类化合物在植物体内含量较低且分离困难,难以满足对其生物和药理活性进行深入研究的需求.
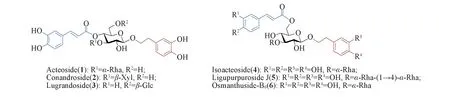
Scheme 1 Some representative phenyl propanoid glycosides
20世纪90年代起,有关苯丙素苷的合成研究工作不断被报道.1998年,Cai等[19]合成了Osmanthuside-B6(6)的2-O-乙酰基衍生物;1999年,Kawada等[20]和van Boom等[21]先后报道了Acteoside(1)的全合成;2000 年和2002 年,Kawada 等[22,23]先后报道了Conandroside(2)和Isoacteoside(4)的全合成.上述工作使用了较多的保护基操作,而且部分保护基与苯丙素苷具有的α,β不饱和酯结构不相容,导致合成效率不高.为降低苯丙素苷类分子的合成路线对保护基的依赖,缩减合成步骤并提高效率,Judeh等[24]在2-氨基乙基二苯基硼酸酯活化下,用卤苷给体与多羟基受体进行区域选择性糖苷化,生成相应的原酸酯,乙酰基保护受体的剩余羟基后,在三氟甲磺酸三甲基硅酯(TMSOTf)促进下转化为1→3连接的二糖,进而完成苯丙素苷Osmanthuside-B6(6)的全合成.
2008 年以来,本课题组[25~30]发展了以糖基邻炔基苯甲酸酯为给体的金(Ⅰ)催化糖苷化反应,该反应的活化机制与经典的糖苷化反应不同,反应条件中性温和,已被应用于多个天然产物的高效合成[31~35].利用葡萄糖硫苷中各个羟基活性的差异,以该方法进行区域选择性糖苷化,可以得到关键的α-(1→3)连接的二糖化合物.在苯丙素苷的合成中发现,该二糖硫苷需要转化成其它给体才能与取代苯乙醇进行糖苷化[36].基于此,本文改进了合成路线,先完成取代苯乙醇葡萄糖苷的合成,再进行区域选择性糖苷化构建α-(1→3)糖连接,并完成了天然苯丙素苷毛蕊花糖苷(Acteoside,1)和异毛蕊花糖苷(Isoacteoside,4)的合成及紫茎女贞苷J(Ligupurpuroside J,5)的首次全合成(Scheme 2).
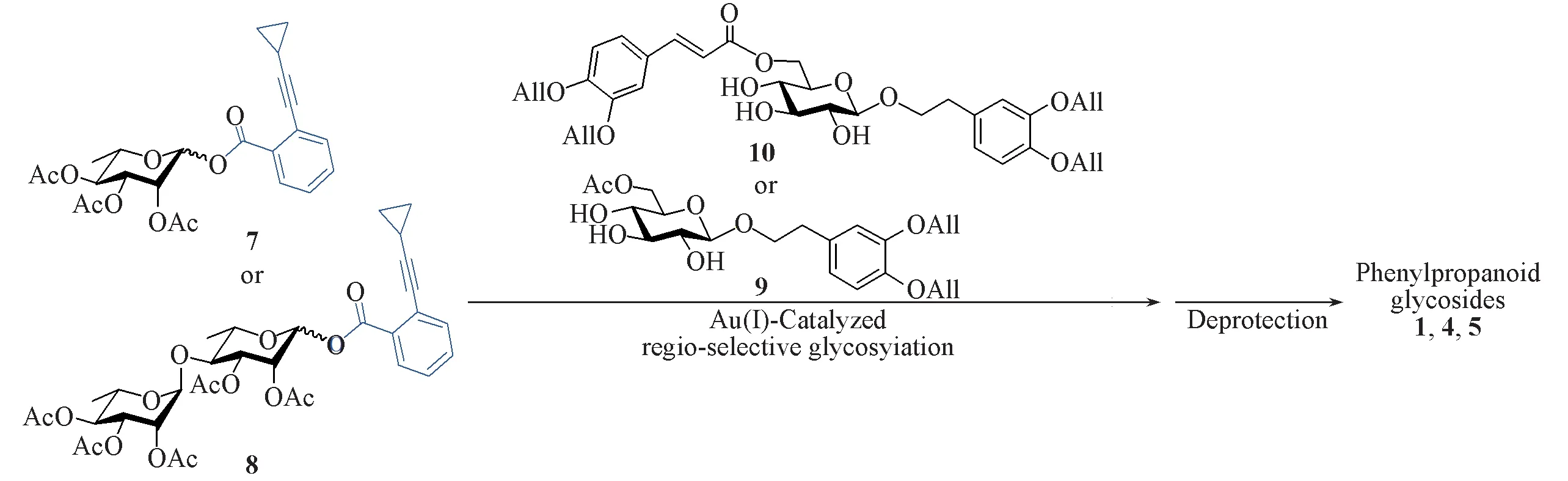
Scheme 2 A new synthetic strategy for phenylpropanoid glycosides
1 实验部分
1.1 试剂与仪器
N-碘代丁二酰亚胺(NIS,纯度98%)、四(三苯基膦)钯(纯度99%)和N,N-二异丙基乙胺(DIPEA,纯度99.5%)购自萨恩化学技术(上海)有限公司;三氟甲磺酸三甲基硅酯(TMSOTf,纯度99%)购自艾览(上海)化工科技有限公司;N-甲基咪唑(NMI,纯度99%)和氯乙酰(纯度98%)购自上海阿拉丁生化科技股份有限公司;乙酸酐(分析纯)购自国药集团化学试剂有限公司;二环己基碳二亚胺(DCC,化学纯)购自上海天莲化工科技有限公司;4-二甲氨基吡啶(DMAP,纯度99%)和质量分数30%~33%甲胺的甲醇溶液购自上海百灵威化学技术有限公司;草酰氯(纯度>98%)购自梯希爱(上海)化成工业发展有限公司;四丁基氟化铵四氢呋喃溶液(1 mol/L)和1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDCI,纯度99%)购自上海毕得医药科技有限公司.常用溶剂采用微波炉活化的0.4 nm 分子筛进行干燥;柱层析所用溶剂均为分析纯.
Agilent-500 型核磁共振波谱仪[NMR,以四甲基硅烷(TMS)作为内标,美国Agilent 公司];Agilent 6224 TOF LC/MS型和Agilent TOF LC/MS 1260-6230型高分辨质谱仪(HRMS,美国Agilent公司);Anton Paar MCP 5500型旋光仪(光源为钠源589 nm,奥地利Anton Paar公司);ZF-Ⅰ型三用紫外分析仪(波长为254 nm,上海豫康科教仪器设备有限公司).
1.2 实验过程
参照文献方法合成了三苯基膦金(I)双(三氟甲磺酰基)亚胺盐(PPh3AuNTf2)和邻环丙基乙炔基苯甲酸[26]、3,4-二-O-烯丙氧基苯乙基-β-D-吡喃葡萄糖苷(11)[37]、2-环丙乙炔基苯甲酰基2,3,4-三-O-乙酰基-L-吡喃鼠李糖苷(7)[25,38]、三氯乙酰亚胺酯基2,3,4-三-O-乙酰基-L-吡喃鼠李糖苷(12)[39]、3,5-二甲基-4-(2′-苯乙炔苯基)苯基2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖苷(13)[40]、3,4-二-O-烯丙氧基-E-肉桂酸(14)[41]和叔丁基二甲基硅基2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→4)-2,3-二-O-异亚丙基-α-L-吡喃鼠李糖苷(15)[40].
1.2.1 3,4-二-O-烯丙氧基苯乙基-6-O-乙酰基-β-D-吡喃葡萄糖苷(9)的合成 将4.6 g(11.6 mmol)化合物11 分散于250 mL 乙酸乙酯中,加入125 μL(2.3 mmol)浓硫酸,于40 ℃反应至用薄层色谱(TLC)监测原料基本消耗完全(约40 h);依次用饱和NaHCO3水溶液和饱和NaCl溶液洗涤,用乙酸乙酯萃取,合并有机相,用无水Na2SO4干燥,经过滤、浓缩、柱层析[V(二氯甲烷)∶V(甲醇)=40∶1至20∶1]得到3.2 g白色固体9,产率为62%,回收产率为68%.
1.2.2 3,4-二-O-烯丙氧基苯乙基-2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→3)-6-O-乙酰基-β-D-吡喃葡萄糖苷(16a)及具有类似结构的副产物16b,16c 和16d 的合成 将137 mg(0.3 mmol)糖苷化给体7 和263 mg(0.6 mmol)受体9溶解于13 mL干燥的二氯甲烷中,加入1 g活化的0.4 nm分子筛,在氩气保护下于室温搅拌反应0.5 h.将反应体系温度降至-60 ℃,加入PPh3AuNTf2的二氯甲烷溶液(0.03 mol/L,1 mL),在-60 ℃下反应10 h;升温至-50 ℃继续反应30 h;升温至室温并加入三乙胺淬灭反应.将反应液垫硅藻土过滤并浓缩,经柱层析[V(石油醚)∶V(乙酸乙酯)=4∶1至2∶1至1∶1],得到白色泡沫状固体16a和16d的混合物共125 mg,其中16a 109 mg,产率为51%,16d 15 mg,产率为10%,另外得到40 mg白色固体16b(产率为19%)和很少量的16c.
1.2.3 3,4-二-O-烯丙氧基苯乙基-2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→3)-2,6-二-O-乙酰基-4-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(17)的合成 将245 mg(0.35 mmol)化合物16a和82 μL(1.0 mmol)NMI 溶解于15 mL 干燥的二氯甲烷中,在氩气保护及冰浴下缓慢加入39 μL,0.4 mmol乙酸酐,于0 ℃反应24 h;升温至室温继续反应15 h,反应液用饱和食盐水洗涤,用乙酸乙酯萃取水相2次,合并有机相,用无水Na2SO4干燥,经过滤、浓缩及快速柱层析[V(石油醚)∶V(乙酸乙酯)=2∶1至1∶1]得2-酰基产物粗品.将该粗品、182 mg(0.7 mmol)化合物14、63 mg(0.5 mmol)DMAP和180 mg(0.9 mmol)DCC溶解于5 mL干燥的二氯甲烷中,于室温反应至原料消耗完全;用乙酸乙酯稀释,垫硅藻土过滤,用饱和NaCl 溶液洗涤,有机相用无水Na2SO4干燥,经过滤、浓缩、柱层析[V(石油醚)∶V(丙酮)=6∶1至4∶1]得到153 mg白色泡沫状固体17,产率为44%.
1.2.4 3,4-二-O-烯丙氧基苯乙基-α-L-吡喃鼠李糖-(1→3)-4-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(18a)的合成 将60 mg(0.06 mmol)化合物17 溶解于3 mL 二氯甲烷中,于-30 ℃加入3 mL 质量分数为30%~33%甲胺的甲醇溶液,保持低温反应5 h.将体系温度升至-25 ℃反应5 h,浓缩后经柱层析[V(二氯甲烷)∶V(甲醇)=40∶1]得到25 mg化合物18a,产率为53%.
1.2.5 Acteoside(1)的合成 将24 mg(30 μmol)化合物18a和52 mg(45 μmol)(PPh3)4Pd溶解于3 mL脱气乙酸中,于60 ℃反应5 h,将体系温度升至70 ℃反应至原料消耗完全.浓缩后用制备板([V(二氯甲烷)∶V(甲醇)∶V(乙酸)=3∶1∶0.1)分离后,经反相C18柱层析[V(水)∶V(甲醇)=4∶1至2∶1]得到14 mg棕黄色固体Acteoside,产率为76%.
1.2.6 3,4-二-O-烯丙氧基苯乙基-6-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(10)的合成 将2.6 g(10.0 mmol)化合物14 溶解于50 mL 甲苯中,在氩气保护下,于0 ℃先后加入15 μLN,N-二甲基甲酰胺和6 mL 草酰氯;保持0 ℃反应0.5 h,将体系温度升至室温继续反应3 h,浓缩,抽真空1 h,备用.将3.3 g(8.32 mmol)化合物11溶解于60 mL干燥的二氯甲烷和5 mL干燥的吡啶中,在氩气保护下,于-10 ℃通过注射器加入10 mL(1.0 mol/L)新制备的酰氯二氯甲烷溶液;于-5 ℃反应10 h后升温至10 ℃继续反应10 h;将体系用乙酸乙酯稀释,依次用稀盐酸(1 mol/L)和饱和NaCl溶液洗涤,用无水Na2SO4干燥,经过滤、浓缩、柱层析[V(石油醚)∶V(乙酸乙酯)=1∶1至1∶2至V(二氯甲烷)∶V(甲醇)=15∶1]得到3.0 g白色固体10,产率为56%,回收产率为63%.
1.2.7 3,4-二-O-烯丙氧基苯乙基-2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→3)-6-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(19a)和3,4-二-O-烯丙氧基苯乙基-2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→2)-6-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(19b)的合成 以 46 mg(0.1 mmol)给体7和127.7 mg(0.2 mmol)受体10为原料,参照类似合成化合物16a的选择性糖苷化条件制得化合物19a,粗品经柱层析[V(石油醚)∶V(乙酸乙酯)=4∶1,2∶1,1∶1]得到50 mg白色固体19a,产率为55%,另外得到20 mg 19b,产率为22%.
1.2.8 3,4-二-O-烯丙氧基苯乙基-α-L-吡喃鼠李糖-(1→3)-6-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(18b)的合成 将45.6 mg(0.05 mmol)化合物19a溶解于5 mL甲醇和1 mL二氯甲烷的混合溶剂中,于0 ℃加入35 μL(0.5 mmol)乙酰氯,逐渐升温至室温反应30 h,用三乙胺淬灭反应,浓缩后经柱层析[V(二氯甲烷)∶V(甲醇)=30∶1]得到29.4 mg 化合物18b,产率为75%.另外,以36 mg(40 μmol)化合物19a为原料,使用甲胺条件脱除乙酰基,参照类似合成化合物18a的方法得到29 mg化合物18b,产率为92%.
1.2.9 Isoacteoside(4)的合成 以20 mg(25 μmol)化合物18b为原料,参照类似18a脱除烯丙基的方法得到12 mg棕黄色固体Isoacteoside,产率为79%.
1.2.10 2-环丙乙炔基苯甲酰基-2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→4)-2,3-二-O-乙酰基-L-吡喃鼠李糖苷(8)的合成 将295 mg(0.5 mmol)化合物15加入10 mL乙酸水溶液(体积分数为80%)中,于50 ℃反应4 h,用乙酸乙酯稀释并用饱和NaHCO3溶液中和,用乙酸乙酯萃取2次,有机相用无水Na2SO4干燥,过滤,浓缩后与甲苯共沸除水2 次得到浅黄色固体;将该固体和61 mg(0.5 mmol)DMAP 溶于5 mL干燥的吡啶中,于0 ℃通过注射器加入0.2 mL(2.0 mmol)乙酸酐;逐渐升温至室温并反应至原料消耗完全(约3 h,TLC监测);加入1 mL甲醇于室温搅拌1 h以消耗多余的乙酸酐,除去大部分溶剂,用乙酸乙酯稀释并依次用稀盐酸(1 mol/L)和饱和NaHCO3溶液洗涤,经无水Na2SO4干燥、过滤、浓缩得到粗品;将粗品溶于10 mL 四氢呋喃中,冰浴下滴加1 mol/L 四丁基氟化铵四氢呋喃溶液,冰浴下反应0.5 h,升温至室温并反应至原料消耗完全;用乙酸乙酯稀释,饱和NaCl 溶液洗涤,用乙酸乙酯萃取2次,有机相用无水Na2SO4干燥,过滤,浓缩后与甲苯共沸除水2次,得到异头位裸露的二糖粗品;将该粗品、121 mg(0.65 mmol)邻环丙基乙炔基苯甲酸,124 mg(0.65 mmol)EDCI,79 mg(0.65 mmol)DMAP和176 μL(1.0 mmol)DIPEA溶解于5 mL干燥的二氯甲烷中,室温下反应至原料消耗完全,用乙酸乙酯稀释,有机相用饱和NaCl溶液洗涤,经无水Na2SO4干燥、过滤、浓缩、柱层析[V(石油醚)∶V(乙酸乙酯)=3∶1]得到244 mg白色泡沫状固体8,产率为71%.
1.2.11 3,4-二-O-烯丙氧基苯乙基-2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→4)-2,3-二-O-乙酰基-α-L-吡喃鼠李糖-(1→3)-6-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(20a)和3,4-二-O-烯丙氧基苯乙基-2,3,4-三-O-乙酰基-α-L-吡喃鼠李糖-(1→4)-2,3-二-O-乙酰基-α-L-吡喃鼠李糖-(1→2)-6-O-(3,4-二-O-烯丙氧基-E-肉桂酰基)-β-D-吡喃葡萄糖苷(20b)的合成 以34.4 mg(0.05 mmol)给体8 和63.9 mg(0.1 mmol)受体10为原料,参照类似合成化合物16a的选择性糖苷化条件制得化合物20,粗品经柱层析[V(石油醚)∶V(乙酸乙酯)=4∶1,2∶1,1∶1]得到32 mg白色固体化合物20a,产率为57%,另外得到12 mg化合物20b,产率为19%.
1.2.12 Ligupurpuroside J(5)的合成 以23 mg(20 μmol)化合物20为原料,参照类似化合物17脱除乙酰基的方法采用甲胺条件脱除乙酰基,得到粗品后,参照类似化合物18a 脱除烯丙基的方法得到9.9 mg棕黄色固体Ligupurpuroside J,产率为64%.
2 结果与讨论
2.1 Acteoside的全合成
参照前文[37]方法制备了烯丙基保护的2-苯乙基-β-D-葡萄糖苷11,其糖环上4个羟基裸露.在催化量浓硫酸条件下化合物11 与乙酸乙酯发生酯交换反应得到6-位羟基选择性乙酰化的糖苷化受体9[42](见Scheme 3).

Scheme 3 Synthetic route for acceptor 9
使用鼠李糖邻炔基苯甲酸酯给体7[25,38]和受体9 在Ph3PAuNTf2催化下进行了区域选择性糖苷化反应的尝试(见表1).首先,反应在-70 ℃下进行,使用10%(摩尔分数)的PPh3AuNTf2作催化剂,二氯甲烷作溶剂,反应12 h 后以13%的产率得到主要的α-(1→3)糖苷化产物16a,同时回收了65%的给体(表1中Entry 1).为提高给体的转化率,升高反应温度至-50 ℃并延长反应时间,以51%的收率得到糖苷化产物16a,同时获得少部分的α-(1→2)连接的二糖16b(19%)和三糖16d(10%)(表1中Entry 2).为了缩短反应时间,进一步升高反应温度至-30 ℃,发现选择性明显下降(表1中Entry 3).改变溶剂为甲苯或乙醚,糖苷化选择性发生逆转,α-(1→2)糖苷化产物16a 成为优势产物(表1中Entries 4和5).将金催化剂替换为新制的PPh3AuOTf,发现糖苷化选择性同样下降(表1中Entry 6).为与其它糖苷化方法作对比,将金(I)催化条件替换为TMSOTf或TMSOTf/N-NIS活化条件,使用三氯乙酰亚胺酯给体12[39]和3,5-二甲基-4-(2′-苯乙炔苯基)苯基(EPP)糖苷给体13[40]分别与受体9在-50 ℃下进行反应,反应的选择性明显降低,同时给体7 的选择性也几乎消失(表1 中Entries 7~9).上述结果证明,Au(I)催化糖苷化反应的条件温和,在低温下可以实现一定的区域选择性.产物中二糖16a和少量的三糖16d极性相似,难以分离,但这并不影响后续反应,再经过2步反应后即可分离除去相应杂质.

Table 1 Regioselective glycosylation of acceptor 9 with donors 7,12 and 13a
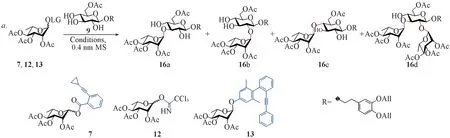
b.Isolated yield;c.the ratio was estimated by TLC;d.NMR yield.
经区域选择性糖苷化反应得到二糖16a后,对葡萄糖2-位进行了选择性保护.由于该位点羟基位阻较大,硅基和氯乙酰基均很难对其进行保护,最后采用体积较小的乙酰基进行保护.通过筛选反应条件发现,使用乙酸酐作为乙酰化试剂,NMI作为碱,室温下可以相对较高的转化率和产率得到2-乙酰化产物,但其氢谱包含2套信号(比例约为5∶1),推测杂质可能为4-乙酰化产物,两者极性一致,用柱层析无法分离;该粗品与烯丙基保护的咖啡酸片段14[41]缩合后得到的化合物17可分离纯化,2步反应的产率为44%.
随后进行了保护基的脱除.计划先脱除烯丙基后脱除乙酰基,但实验中发现在甲胺条件下脱除乙酰基时会产生较多副产物,并且与终产物极性相似,可以是咖啡酸支链断裂的产物,使用反相C18柱层析分离困难.因此,尝试先乙酰基后烯丙基的脱除顺序,在乙酰基的脱除中,起初在反应体系中观察2个主点.因为葡萄糖2-位位阻较大,保护基较难脱除,假设极性较小的产物为葡萄糖2-位乙酰基未脱除产物,补加甲胺溶液使之向极性较大的产物转化,但通过核磁共振氢谱分析发现,这2种产物均无乙酰基的信号,极性较小的化合物为正常脱乙酰基产物18a,同时反应过程中部分底物发生了C4→C6酰基迁移,得到了化合物18b.该化合物脱除烯丙基后可得到另一种苯丙素苷天然产物Isoacteoside.类似条件下的酰基迁移现象已有相关文献报道[23].最后,通过调整甲胺溶液与二氯甲烷的比例以及反应温度,以53%的收率得到产物18a.化合物18a在四(三苯基膦)钯[(PPh3)4Pd]条件下脱除烯丙基,以76%的产率得到目标分子Acteoside(见Scheme 4).
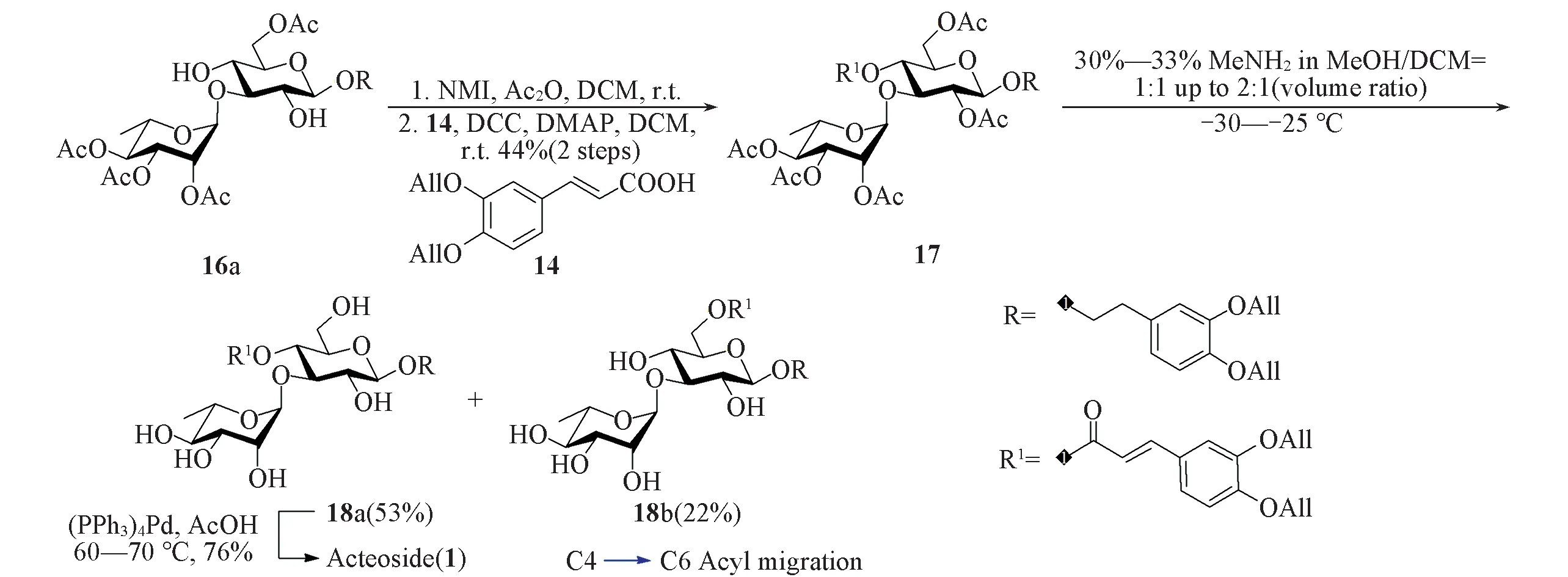
Scheme 4 Synthetic routes for Acteoside(1)
2.2 Isoacteoside和Ligupurpuroside J的合成
如果能进行选择性糖苷化得到所需的α-(1→3)连接二糖,则对于取代肉桂酸支链连接在6-位的苯丙素苷的合成不需要选择性保护2-,4-位羟基.因此,本文对苯丙素苷Isoacteoside(4)[4~6]的合成进行了尝试.以甲苯(Tol)作溶剂,在N,N-二甲基甲酰胺(DMF)的促进下,将烯丙基保护的咖啡酸14制备成相应的酰氯,选择性酰化化合物11 中葡萄糖上6-位伯羟基得到下一步糖苷化所需的受体10(见Scheme 5).
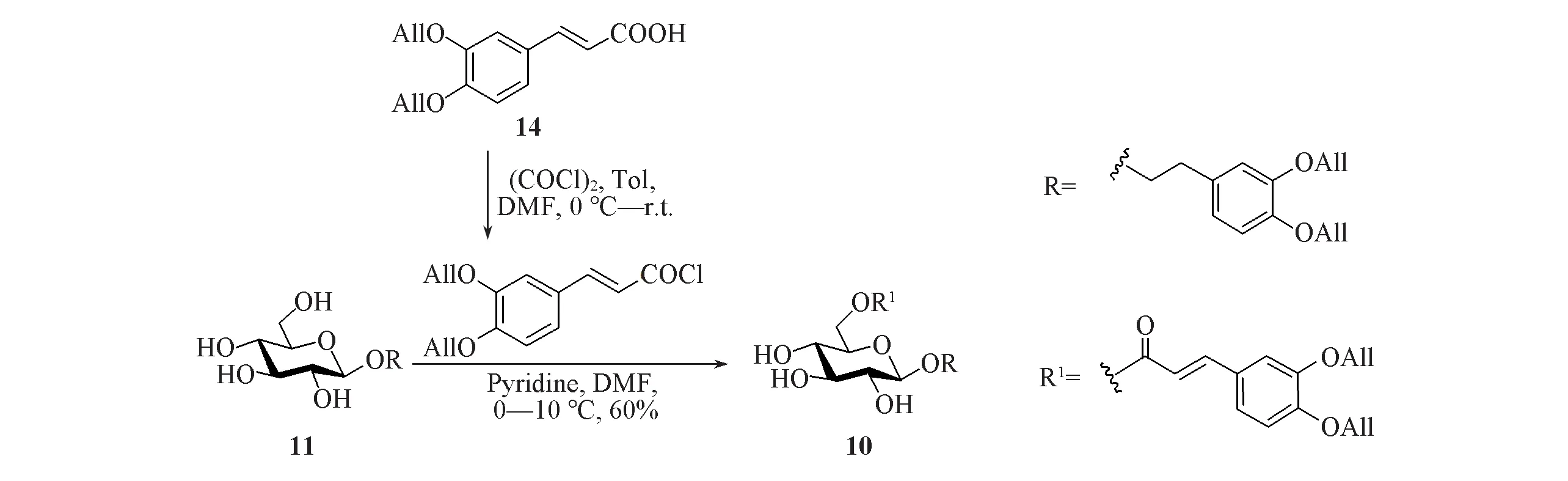
Scheme 5 Synthetic routes for acceptor 10
尝试使用鼠李糖给体7与受体10在优化的条件下进行了区域选择性糖苷化反应.使用2倍摩尔量受体时,以55%的收率得到α-(1→3)糖苷化产物19a,同时分离得到22%的α-(1→2)二糖19b.随后,对化合物19a进行保护基的脱除.脱除乙酰基时,首先尝试了乙酰氯/甲醇条件,以75%的产率得到化合物18b;其次,尝试采用甲胺条件,保持反应温度在-25~-20 ℃,则可以92%的产率高效脱除全部乙酰基.最后,将化合物18a 脱除烯丙基,以79%的产率顺利得到天然产物Isoacteoside[Scheme 6(A)].
将该路线应用到Ligupurpuroside J[7]的合成上.首先,使用已知二糖15[40]为原料,脱除2-,3-位的丙叉保护基,再用乙酰基保护,随后脱除叔丁基二甲基硅基裸露出异头位羟基,制得二糖邻炔基苯甲酸给体8.给体8与受体10进行区域选择性糖苷化反应,以57%的收率得到所需α-(1→3)三糖20a,另外,也得到了少部分的α-(1→2)三糖20b(19%).对化合物20a采用类似的脱保护基操作脱除乙酰基和烯丙基,得到了苯丙素苷Ligupurpuroside J[见Scheme 6(B)].
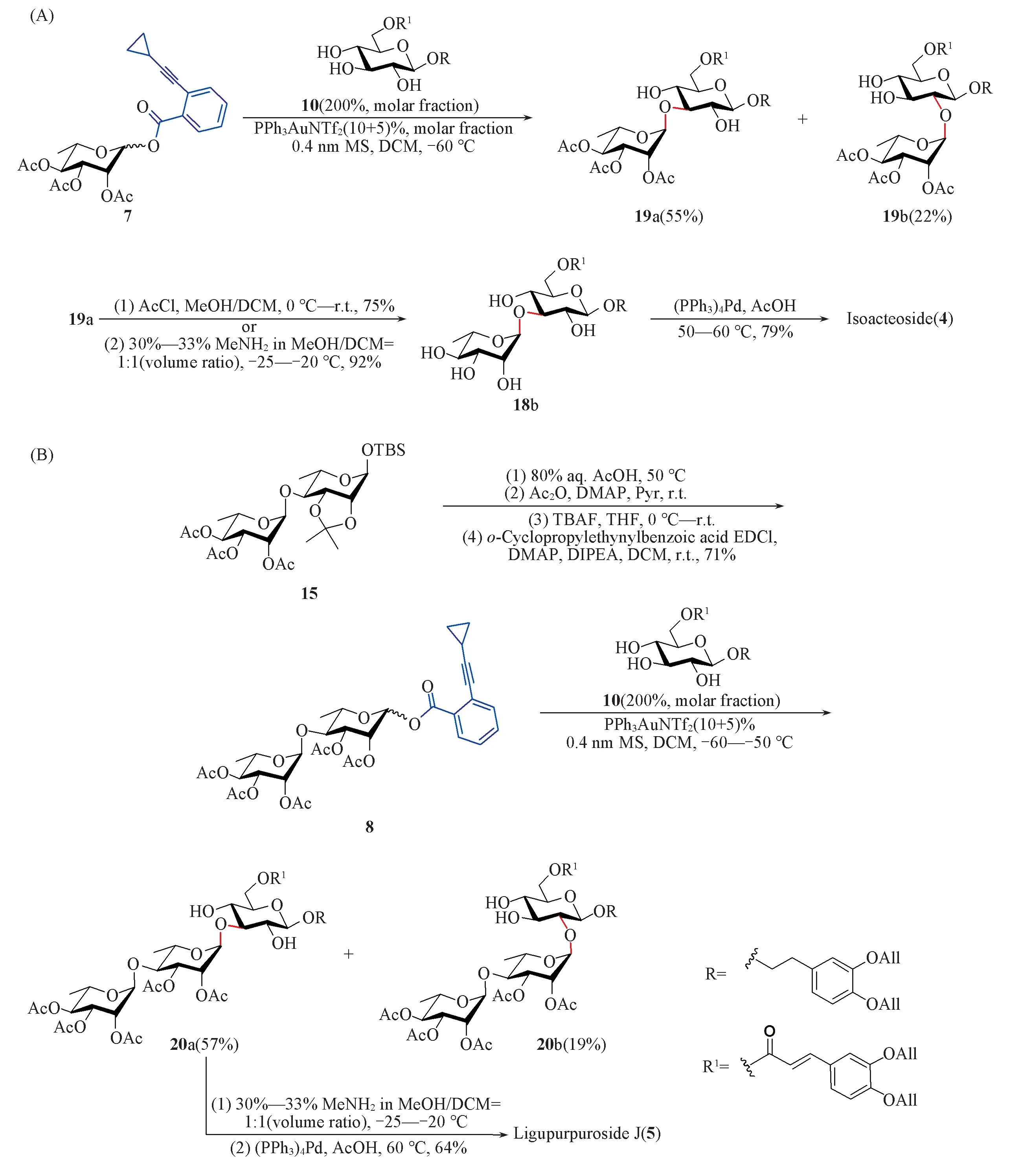
Scheme 6 Synthetic routes for Isoacteoside and Ligupurpuroside J
化合物8~10,16~20,Acteoside(1),Isoacteoside(4)和Ligupurpuroside J(5)的比旋光度和高分辨质谱数据列于表2,核磁共振氢谱和碳谱的数据列于表3,核磁共振的原始谱图见图S1~图S36(见本文支持信息).

Table 2 Optical rotation values and HRMS data of compounds 8—10,16—20,Acteoside(1),Isoacteoside(4)and Ligupurpuroside J(5)
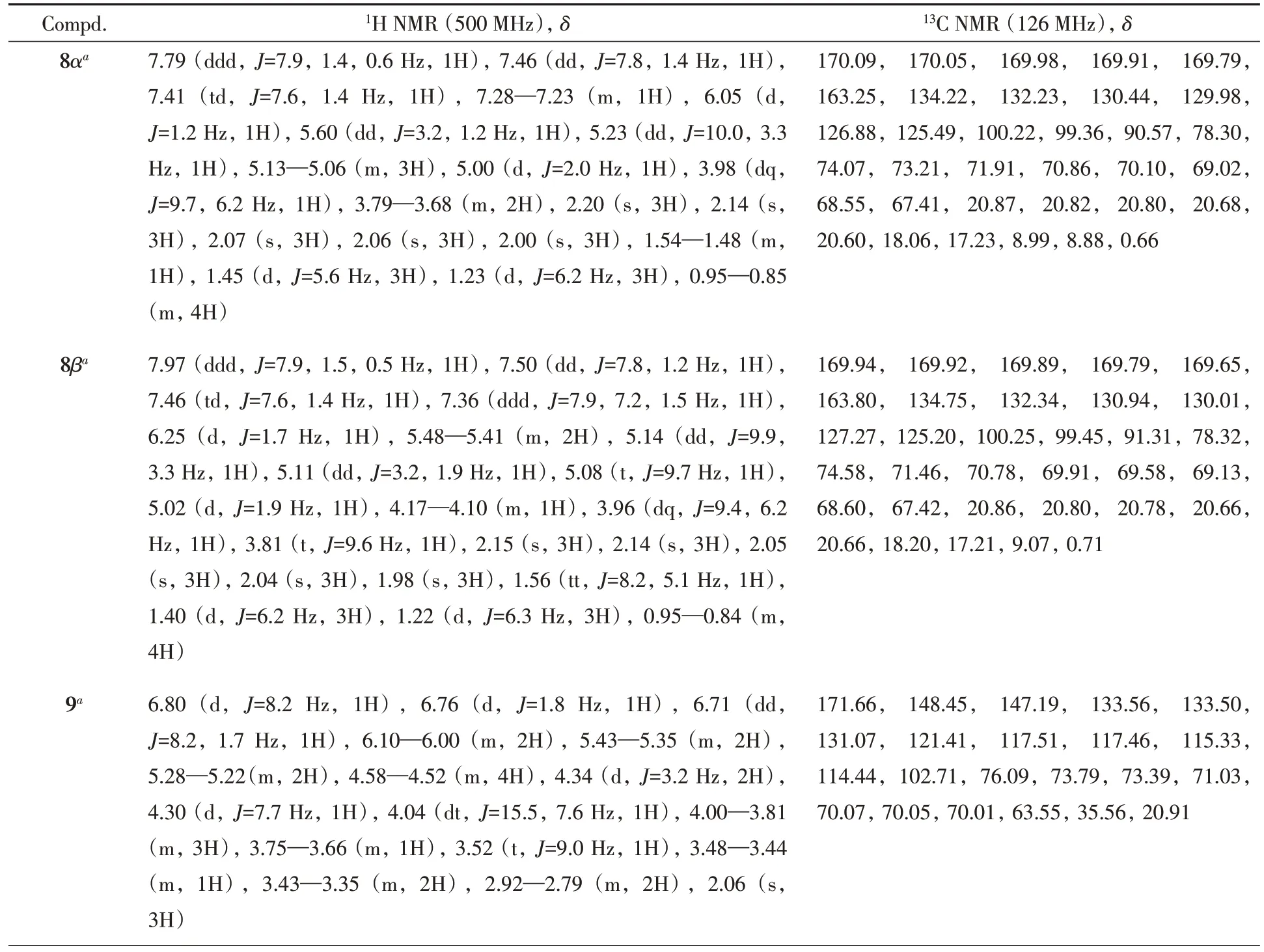
Table 3 1H and 13C NMR data of compounds 8—10,16—20,Acteoside(1),Isoacteoside(4)and Ligupurpuroside J(5)

Continued
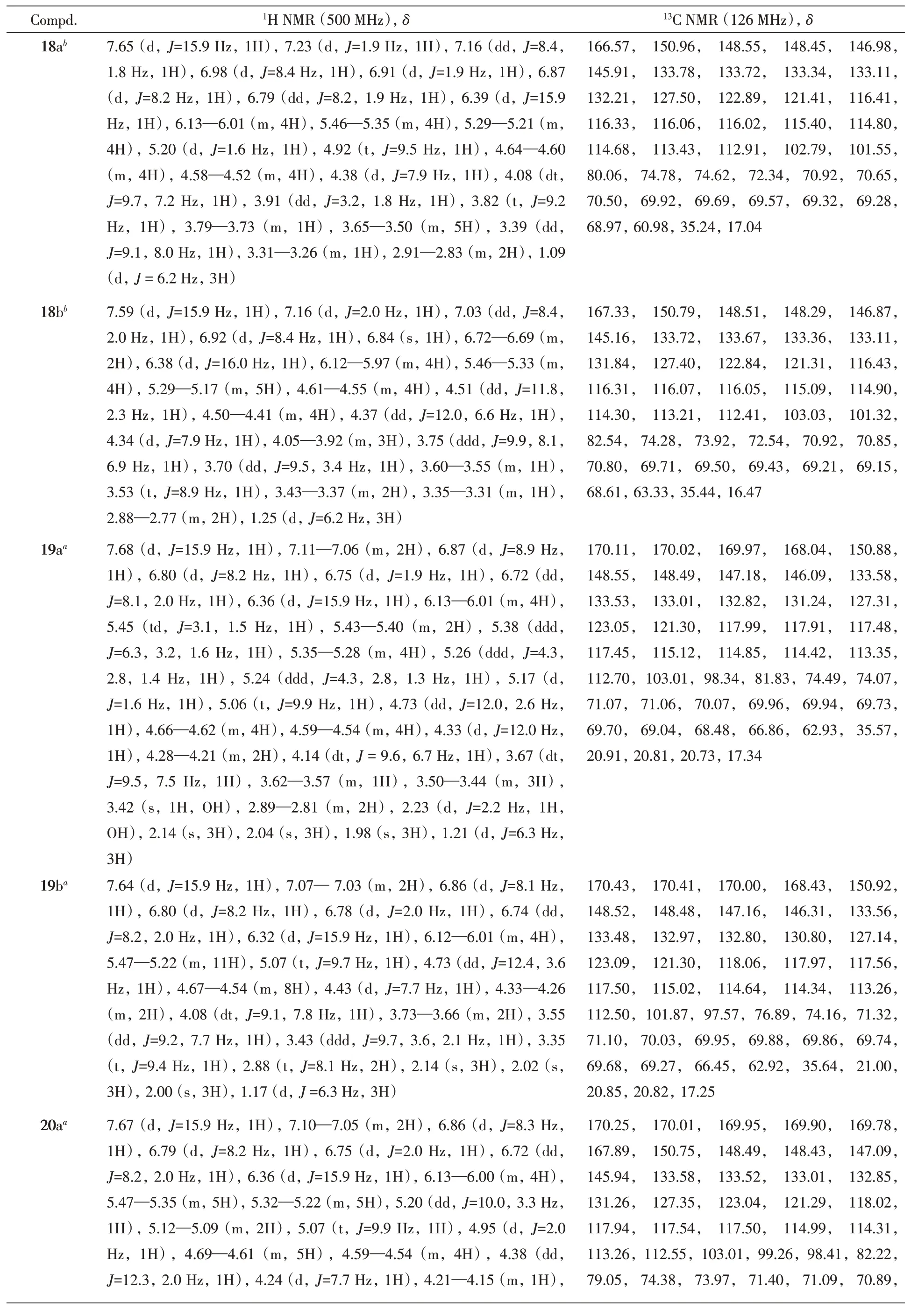
Continued
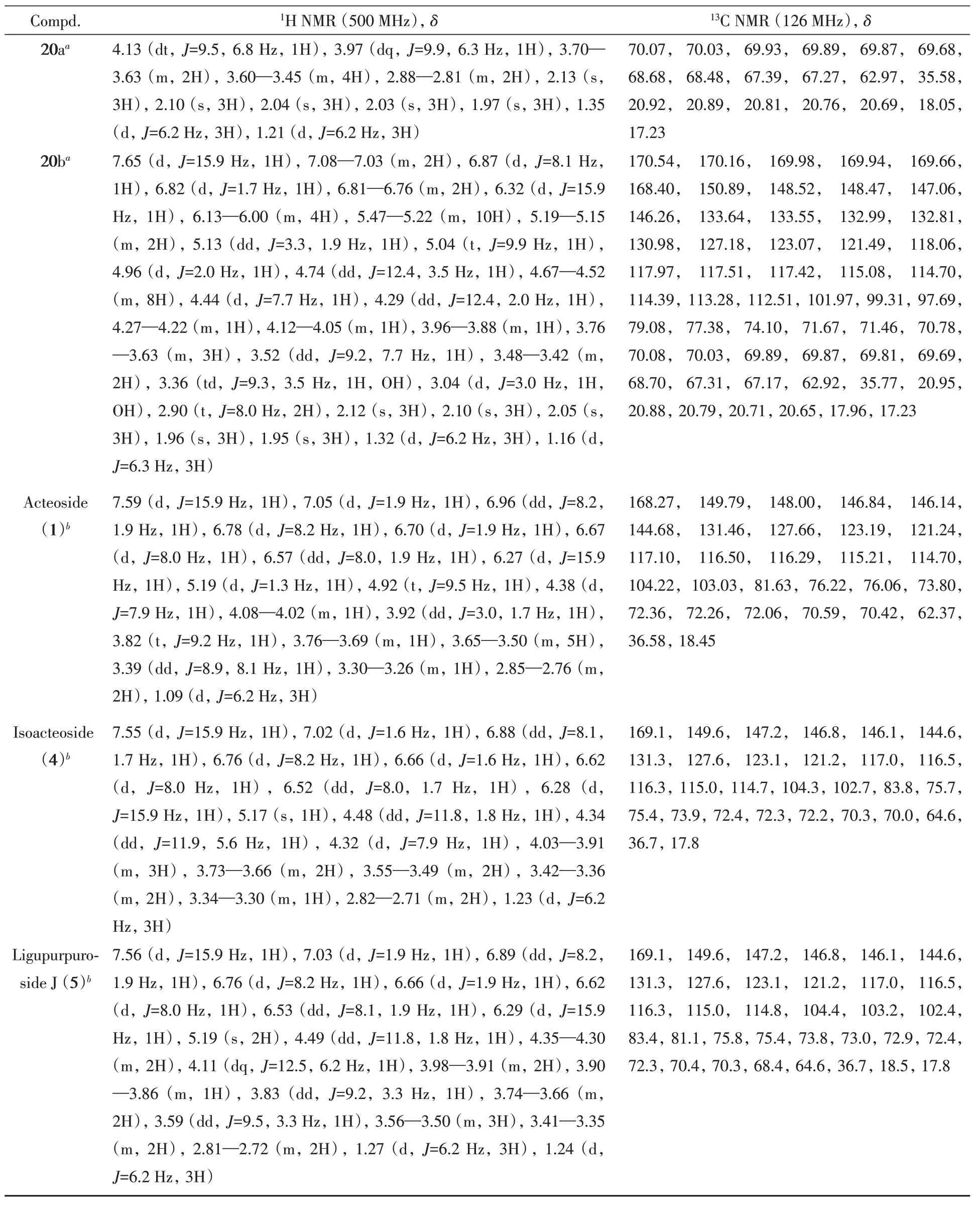
Continued
3 结 论
采用条件温和的金(Ⅰ)催化糖苷化方法,区域选择性地构建苯丙素苷中常见的α-(1→3)糖苷键,缩短了合成路线并提高了合成效率.以此为关键反应,从已知化合物11出发,分别以6步反应6%的收率,4 步反应24%的收率完成了苯丙素苷Acteoside(1)和Isoacteoside(4)的合成;从已知二糖15 出发,以7步反应26%的收率完成了Ligupurpuroside J(5)的首次全合成.这些化合物的合成验证了该苯丙素苷合成路线的通用性.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20200266.
猜你喜欢
陕西师范大学学报(自然科学版)(2022年5期)2022-11-09
化学工程师(2022年5期)2022-05-11
云南化工(2020年4期)2020-05-19
科海故事博览·中旬刊(2020年3期)2020-03-15
安徽化工(2018年5期)2018-10-23
天然产物研究与开发(2018年8期)2018-09-10
烟草科技(2015年8期)2015-12-20
世界热带农业信息(2014年10期)2014-11-13
天然产物研究与开发(2014年6期)2014-04-27
影像科学与光化学(2014年3期)2014-03-11
