
腓骨肌萎缩症4B3型一家系的临床及基因分析
2020-10-23 06:53颉满珍王根绪
中国实用神经疾病杂志 2020年19期
颉满珍 王根绪
天水市第一人民医院,甘肃 天水 741000
腓骨肌萎缩症是遗传性运动性感觉神经病,又称夏科-马利-杜斯病(Charcot-Marie-Tooth disease,CMT),是神经系统疾病中最常见的遗传性周围神经病之一,也是人类最常见的遗传性疾病之一[1],具有高度遗传和临床异质性[2],在不同人群中CMT的发病率为(17~40)/100 000[3-5]。全世界已有较多的相关病例,对这些病例进行详细分析后发现大多数患者属于遗传性周围神经病,发病主要在儿童期至青少年,30岁以前发病者占78.2%,40岁以后发病者仅占8.1%,平均发病年龄19.2岁[6]。CMT是以慢性进行性四肢远端无力、肌肉萎缩、双下肢腱反射消失、共济失调和末梢循环差为主,伴弓形足、脊柱侧弯等骨骼畸形为临床表现形式的综合性疾病[7]。该病主要呈慢性进行性发展,早期出现肌无力,活动不耐受,后逐渐发展为双下肢肌肉萎缩、骨骼畸形[8-9],导致双下肢活动无力,生活不能自理。在根据疾病的临床表现、家族史、神经神经电生理及病理特点的基础上考虑可能的相关疾病,再进行基因突变分析,60%~70%的CMT患者能通过基因检测而明确诊断,为疾病治疗、预后的判断、遗传咨询等方面提供指导性意见[10-11]。CMT的临床表现、分型、神经电生理及致病基因等方面的研究有了令人瞩目的成就,不仅对以往的难题作出合理的解释,也发现了一些新的挑战。随着研究的不断深入,新型CMT逐渐被发现,但其分型却越来越复杂。根据目前的研究结果,CMT的遗传方式主要呈X连锁显性(XD)或隐性遗传(XR)、常染色体显性(AD)及常染色体隐性(AR),尤其以AD遗传为主要方式[12],而已知分型有CMT1、CMT2、CMT3、CMT4、CMT5、CMT6、CMTDI、CMTRI、CMTX等[13]。本文通过调查腓骨肌萎缩症4B3型患者一家系,进一步研究其主要临床表现、神经肌电图和基因突变特点。
1 病例资料
1.1临床表现及辅助检查先证者为34岁女性,系甘肃省陇南市人,长期在家务农,主要以“进行性双下肢无力、肌肉萎缩10余年,加重6个月”于2018-04-10收住天水市第一人民医院神经内科。患者于入院前10余年无明显诱因出现双足行走无力,主要表现为双下肢乏力,伴小腿肌肉酸痛,尤其在长时间活动后明显,休息后好转,后症状逐渐加重,逐渐出现双下肢活动无力,行走不能,需家属搀扶,生活不能完全自理。神经系统体格检查:意识清楚,言语清晰,对答清晰,定向力、智力、计算力正常,十二对脑神经检查未见明显异常。双上肢肌容、肌张力、肌力基本正常,双下肢腓肠肌重度萎缩,呈“倒酒瓶征”,肌张力降低,双下肢胫前肌2级,腓肠肌肌力1级,双足背伸不能,跖屈活动受限,双膝关节以下深浅感觉均减弱,尤其远端较重,膝腱反射减弱,跟腱反射基本消失,双侧病理征阴性。实验室检查:生化检查示肌酸磷酸激酶(CPK)和乳酸脱氢酶(LDH)增高。脑脊液检查示生化、细胞数基本正常;肌电图检查:双正中神经和尺神经感觉传导速度及波幅基本正常,双侧胫前肌插入电位时限延长,静息时可见自发电位;轻度收缩时波幅较前增高,动作电位时限较前延长,而用力收缩时呈单纯相。双侧腓总神经在强刺激时动作电位、波幅均未引出。头颅MRI示双侧半卵圆中心周围散在脱髓鞘,其余未见明显异常;全脊柱MRI可见部分颈椎椎间盘轻度膨出,其余未见明显异常。
1.2家系调查见图1。调查此家系婚育史,在三代以内均未发现近亲结婚史,Ⅳ1、Ⅳ2、Ⅳ3、Ⅳ7均出现不同程度的活动不耐受或肌无力、肌萎缩,经患者及家属知情同意后,遂对其4人(Ⅲ2、Ⅳ1、Ⅳ2、Ⅳ3)进行基因检测,发现4人均出现腓骨肌萎缩相关基因突变;Ⅰ4(已死亡),根据家属描述,自幼发病(具体不详),缓慢出现双下肢无力,中年后生活不能自理,死因不详;Ⅱ5(已死亡),据家属述说,自10岁左右出现双下肢活动无力,20岁左右需要拄拐杖才能行走,30岁死于肺炎;Ⅲ2(先证者),约17岁发病,起初发病时双下肢活动乏力,后逐渐加重,出现走路困难,拄拐杖才能行走;Ⅳ1,年龄8岁,在5岁感行走乏力,其余无特殊不适。Ⅳ2,4岁儿童,目前未出现肢体无力或活动不耐受等症状;Ⅳ3,3岁儿童,目前年龄较小,未出现活动不耐受等症状。Ⅳ7(未行基因检测),20岁,15岁发病,主要为双下肢乏力,活动不耐受,有双下肢麻木感。
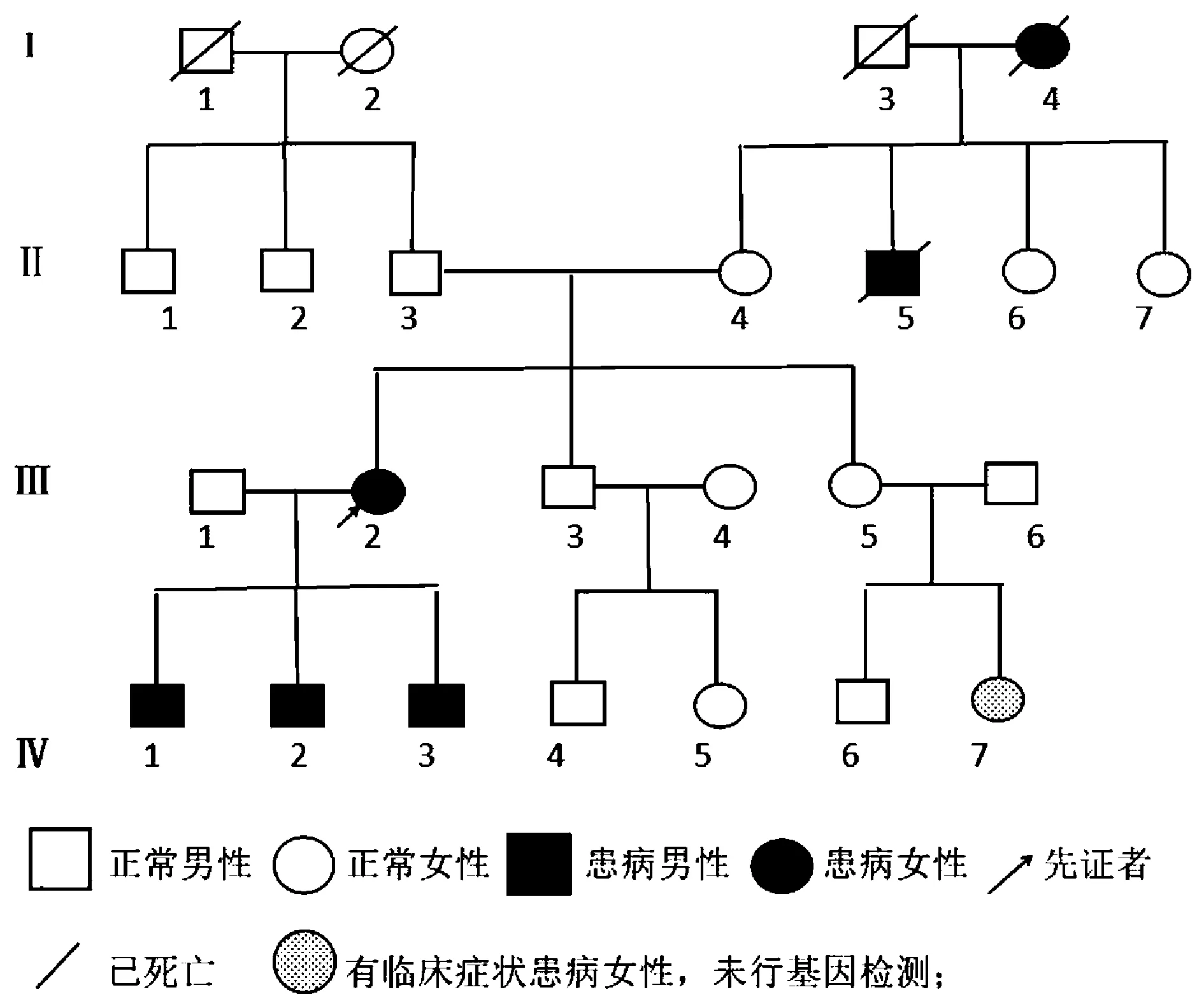
图1 CMT4B3型患者家系系谱图Figure 1 CMT4B3 pedigree of patients
2 讨论
2.1腓骨肌萎缩症的命名及分型目前,腓骨肌萎缩症的分类主要还是根据临床表现、神经电生理检查结果、神经肌肉活检、遗传方式、基因检测。根据神经电生理及病理特点,可以将该疾病分为脱髓鞘型(CMT1型)、轴突型(CMT2型)、轴索变性和脱髓鞘共存的中间型(ICMT型)[14]。近年来,AR-CMT型被命名为CMT4型,X连锁遗传(显性及隐性)CMT型被命名为CMTX型[15]。研究发现部分中间型AD-CMT病例在电生理检查中发现既有神经元轴突变性,也有神经脱髓鞘,因此该型也被命名为中间型常染色体显性遗传CMT(dominant intermediate CMT,DI-CMT)[16]。根据神经电生理检查,脱髓鞘型患者的NCV显著降低,上肢运动神经的神经传导速度多在38 m/s以下,而轴突型患者一般仅轻度降低或正常,DI-CMT和CMTX型患者的神经传导速度(nerve conduction velocities,NCV)一般波动在CMT1和CMT2之间,介于25~45 m/s[17]。而腓肠肌的神经电生理检查对CMT的诊断具有重要的价值[18],神经肌肉活检一般显示有髓神经纤维的数量明显减少,脱髓鞘,部分髓鞘壁增厚,髓鞘破坏明显,无髓神经纤维肿胀,胶原纤维增生,形成“洋葱头”样改变[19-21]。
根据流行病学调查,腓骨肌萎缩症在地中海沿岸国家较为常见,且存在表型异质性。主要临床特点除常见的表现外,还有一些特征性症状,如大多数幼年患者呈慢性进行性发展,从而导致双下肢远端肢体畸形,丧失行走能力[22]。也有少数患者出现脑神经损害[23]、早发、白内障、青光眼、听力下降、无精、呼吸肌麻痹及声带麻痹[24-25]等表现。CMT4型以常染色体隐性遗传为主,在近亲结婚的家系中其后代发病率相对较高。目前发现的受累基因共11种,除HMNSR外,10型疾病基因均被克隆。根据受累基因的不同,可将其分为11种分型,CMT4A、4B1、4B2、4B3、4C、4D、 4E、4F、4G、4H和4J[26]。目前根据临床表现形式、神经电生理检查仍无法详细区分CMT4的各个亚型,为进一步明确诊断,仍主要依赖于基因突变分析结果。而腓骨肌萎缩症4B型是比较严重且罕见的分型,呈常染色体隐性遗传,根据其受累基因,可将CMT4B型分为3种亚型,第一种为MTMR2基因突变导致的CMT4B1型[27],第二种为MTMR13/SBF2基因突变导致的CMT4B2型,第三种为SBF1基因突变导致的CMT4B3型[28]。
2.2CMT4B3型的分子遗传学研究本家系共4代24人,其中7例出现临床表现,男女均患病,但由于样本较少,男女比例无明显特异性;主要临床表现为进行性双下肢远端乏力,后症状逐渐加重,出现双上肢远端轻度肌肉萎缩,并伴针刺觉减退、弓形足。肌电图检查显示大多数患者存在感觉或运动神经不同程度的减弱或消失,但感觉神经的异常率明显高于运动神经,且感觉神经功能受损的严重程度明显重于运动神经功能;7例患者中仅4例进行了腓骨肌萎缩症基因筛查,其腓骨肌萎缩相关基因均发生突变,在基因组中与腓骨肌萎缩相关的基因SBF1基因有位点发生杂合变异,主要相关疾病为常染色体隐性遗传4B3型(图2)。通过检查本家系成员发现携带者不发病。先证者SBF1基因在chr22:50899958位置上发生碱基C>A的杂合变异,导致编码的氨基酸由脯氨酸变异为苏氨酸,而该位点变异可能有害。基因检测时也发现该患者大儿子该基因位点未发现碱基变异,而第二个儿子、第三个儿子在在SBF1基因rs757639561位点发生杂合变异,同时在先证者SBF1基因的chr22:5090098位置上发生碱基G>A的杂合变异,而家系验证时发现受检者大儿子在SBF1基因rs779542104位点发生杂合变异,而第二个儿子、第三儿子均未发现基因变异。先证者SBF1基因在chr22:50900983 剪切位置上发生碱基G>A的杂合变异,该变异在EXAC数据库东亚人群中变异频率为0.07%,但根据现有数据未发现有明显临床意义。

注:黑色箭头所示为SBF1基因突变发生位点;A:先证者SBF1基因存在复合杂合突变,c.2833C>A(左)和c.2127G>A(右);B:先证者大儿子SBF1基因存在杂合突变,左图正常,c.2127G>A(右);C:先证者二儿子SBF1基因存在杂合突变,c.2833C>A(左),右图正常;D:先证者三儿子SBF1基因存在杂合突变,c.2833C>A(左),右图正常图2 CMT患者及儿子SBF1基因测序Figure 2 CMT patient and son SBF1 gen sequencing
CMT 4B3型是由NAKHRO等[28]通过对一个亚洲国家家系调查,通过基因检测发现,在全外显子组测中确定并将其命名。通过外显子基因序列发现其致病基因为SBF1,其上的杂合突变导致其发病。SBF1/MTMR5在基因结构上与SBF2/MTMR2相类似,同时SBF1也和其他肌管相关蛋白具有相类似的功能,且与内涵体的运输有关。SBF1与MTMR2通过卷曲螺旋结构域的相互作用,影响其亚细胞的定位,增加MTMR2的酶活性,从而在运动神经细胞中发挥重要作用。该家系调查中发现,患病的7例发病年龄8~15岁,起初表现均为双下肢远端的肌乏力,活动后明显,后呈进行性发展,到中年以后肢体远端肌肉萎缩、关节变形,最终丧失生活能力。
目前暂无根治腓骨肌萎缩症4B3型的相关药物,只能进行个体化治疗,可给予营养神经、大剂量B簇维生素、改善循环、活血化瘀等对症支持治疗,再辅以肢体的康复锻炼、中医理疗及外科矫形手术,通过改善患者的运动能力,从而改变其生活质量。最近的一项研究表明,与其他神经肌肉疾病相反,建议在尽可能的情况下进行有氧运动[29]。虽然有些家属不相信康复治疗有明显受益,但腓骨肌萎缩症患者能从康复锻炼中得到身心治疗[30-31]。由于此病发病较隐匿,病程进展较为缓慢,致残程度轻重不一,且容易出现漏诊、误诊,尤其在临床诊疗中如发现四肢远端肌肉萎缩,家系调查时发现一些和该患者具有相似临床症状者,可通过基因筛查明确诊断[32]。
利益冲突:所有作者均声明不存在利益冲突
猜你喜欢
保健与生活(2022年13期)2022-07-06
临床输血与检验(2022年3期)2022-06-22
疯狂英语·新阅版(2021年8期)2021-09-10
种子(2021年3期)2021-04-12
诊断学(理论与实践)(2020年1期)2020-04-28
郑州大学学报(医学版)(2019年3期)2019-06-03
北京广播电视报(2019年8期)2019-03-27
武警医学(2019年2期)2019-03-05
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
