
HPLC-DAD法同时测定人参再造丸中11个成分的含量
2020-11-02 07:40刘惠军
实用药物与临床 2020年10期
张 博,刘惠军,田 兰,王 彦,陈 睿,闫 超*
0 引言
人参再造丸收载于《中国药典》2015年版一部,该方由人参、黄芪等56味药组成,功能益气养血,祛风化痰,活血通络[1]。现代研究表明,人参再造丸在治疗中风和康复阶段取得了很好的疗效[2-3]。方中人参、甘草、黄芪补气以生血;玄参、何首乌、葛根养阴血,生津液,益精血;赤芍活血止痛;防风祛风通络;橘红化痰利气;青皮疏肝化滞,三七、骨碎补活血祛瘀。由于人参再造丸成分复杂,含量测定干扰多,提取分离难度大,现行法定标准《中国药典》2015年版一部仅选用了黄连中的盐酸小檗碱进行定量控制,而未选取君药成分作为质控指标,不能有效控制成药质量[4]。文献中对人参再造丸质量控制方面的研究报道也很少[5-7],本试验建立了本方中11个成分的含量测定方法,首选了君药并兼顾了臣佐使药的代表性物质,覆盖了该方中上述起主要功效的12味主要药材,为人参再造丸更好的质量控制提供了实验依据。
本实验根据中国药典对人参、三七、黄芪、玄参、制何首乌、葛根、赤芍、防风、橘红、青皮、骨碎补、甘草等12种药材指标性成分的含量测定[8],并结合相关文献报道[9-13],拟选取人参再造丸方中以下药材中的指标性成分进行测定:人参和三七(人参皂苷Rg1、人参皂苷Re)、黄芪(毛蕊异黄酮葡萄糖苷)、玄参(哈巴俄苷)、制何首乌(2,3,5,4′-四羟基二苯乙烯-2-O-β-D葡萄糖苷)、葛根(葛根素)、赤芍(芍药苷)、防风(5-O-甲基维斯阿米醇苷)、橘红(橙皮苷)、青皮(橙皮苷、柚皮苷)、骨碎补(柚皮苷)、甘草(甘草苷),综合上述各化合物的性质,设计提取分离手段,采用HPLC-DAD法同时进行定量分析。
1 仪器与试药
1.1 实验仪器 Agilent 1260高效液相色谱仪(包括真空脱气机、四元泵、自动进样器、二极管阵列检测器);KQ5200E超声波清洗器(昆山市超声仪器有效公司);Thermo GP20水浴锅;BUCHI R-215旋转蒸发仪;CPA225D型电子天平(Sartorius)。
1.2 样品与试药 人参再造丸(6批)分别来自市面上常见的4家公司:厂家A (批号:160602),厂家B (批号:20160802),厂家C (批号:15011174、15011175、15011176),厂家D (批号:QB2027)。葛根素(批号:110752-201615,含量:95.4%)、芍药苷(批号:110736-201539,含量:96.4%)、毛蕊异黄酮葡萄糖苷(批号:111920-201304,含量:98.3%)、甘草苷(批号:111610-201005,含量:94.9%)、2,3,5,4′-四羟基二苯乙烯-2-O-β-D葡萄糖苷(批号:110844-201512,含量:91.0%)、5-O-甲基维斯阿米醇苷(批号:111523-201208,含量:96.4%)、柚皮苷(批号:110722-201613,含量:94.3%)、橙皮苷(批号:110721-201617,含量:96.1%)、人参皂苷Rg1(批号:110703-201530,含量:91.7%)、人参皂苷Re (批号:110754-201525,含量:92.3%)、哈巴俄苷(批号:111730-201307,含量:97.1%),以上对照品均购自于中检院。乙腈、磷酸均为色谱纯(Merck),超纯水由Milli Q超纯水机制得,石油醚、甲醇等试剂为分析纯。
2 方法与结果
2.1 溶液的制备
2.1.1 混合对照品溶液的设备 分别精密称取葛根素、芍药苷、毛蕊异黄酮葡萄糖苷、甘草苷、2,3,5,4′-四羟基二苯乙烯-2-O-β-D葡萄糖苷、5-O-甲基维斯阿米醇苷、柚皮苷、橙皮苷、人参皂苷Rg1、人参皂苷Re、哈巴俄苷的对照品适量,加甲醇溶解并稀释,制成质量浓度分别为43.540 56、51.940 32、21.449 06、20.934 94、20.438 6、6.159 96、43.000 8、218.339 2、64.116 64、87.352 72、10.855 78 μg/ml的混合对照品储备溶液,置于4 ℃冰箱避光保存。
2.1.2 供试品溶液的制备 避光操作。取本品10丸(每丸重3 g),剪碎,混匀,精密称定2 g,加硅藻土2 g,混匀后,置索氏提取器中,加石油醚(60~90 ℃)适量,加热回流提取2 h,弃去石油醚液,药渣挥干,加入甲醇100 ml,加热回流提取3 h,提取液回收甲醇并浓缩至近干,残渣加水30 ml微热超声使溶解,通过经预处理的D101型大孔吸附树脂柱(内径为1.5 cm,柱高为12 cm),用水100 ml洗脱,弃去水液,再用80%乙醇液200 ml洗脱,收集续洗脱液,减压回收溶剂至干,残渣加甲醇溶解并转移至20 ml量瓶中,加甲醇稀释至刻度,摇匀,用0.45 μm微孔滤膜滤过,取续滤液,即得。
2.1.3 阴性样品溶液的制备 按工艺方法制备缺少人参、三七、黄芪、玄参、何首乌、葛根、赤芍、防风、橘红、青皮、骨碎补、甘草的阴性样品,并按“2.1.2”项下的方法,制备阴性样品溶液。
2.2 色谱条件 采用Merck Purospher®STAR C18(250 mm×4.6 mm,5 μm)色谱柱,以乙腈(A)-0.05%磷酸水溶液(B)为流动相,梯度洗脱(0~8 min,13.5%A,86.5%B;8~40 min,13.5%A→14%A,86.5%B→86%B;40~82 min,14%A→19%A,86%B→81%B;82~90 min,19%A→20%A,81%B→80%B;90~123 min,20%A→29%A,80%B→71%B);流速1.0 ml/min;柱温35 ℃;进样量:20 μl;检测波长为203 nm(人参皂苷Rg1、人参皂苷Re)、237 nm(芍药苷、5-O-甲基维斯阿米醇苷)、254 nm(葛根素、毛蕊异黄酮葡萄糖苷)、280 nm(甘草苷、柚皮苷、橙皮苷、哈巴俄苷)、320 nm(2,3,5,4′-四羟基二苯乙烯-2-O-β-D葡萄糖苷),理论板数均不得低于10 000。在203 nm的色谱图见图1。
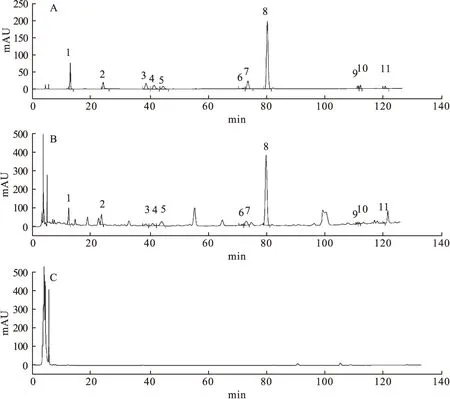
图1 高效液相色谱图注:1.葛根素,2.芍药苷,3.毛蕊异黄酮葡萄糖苷,4.甘草苷,5.2,3,5,4′-四羟基二苯乙烯-2-O-β-D葡萄糖苷,6.5-O-甲基维斯阿米醇苷,7.柚皮苷,8.橙皮苷,9.人参皂苷Rg1,10.人参皂苷Re,11.哈巴俄苷;A.对照品混合溶液,B.人参再造丸样品,C.阴性样品
2.3 线性关系考察 分别精密吸取“2.1.1”项下11种混合对照品储备溶液0.5、1、2、5、10、15、20 ml至20 ml容量瓶中,加甲醇定容,即得不同浓度的11种混合对照品溶液。
按“2.2”项下色谱条件,依次注入液相色谱仪进行测定,以各个对照品进样量(μg)为X轴,峰面积值为Y轴计算回归方程,结果见表1。结果表明,葛根素、芍药苷、毛蕊异黄酮葡萄糖苷、甘草苷、2,3,5,4′-四羟基二苯乙烯-2-O-β-D葡萄糖苷、5-O-甲基维斯阿米醇苷、柚皮苷、橙皮苷、人参皂苷Rg1、人参皂苷Re、哈巴俄苷在各自浓度范围内线性关系良好。
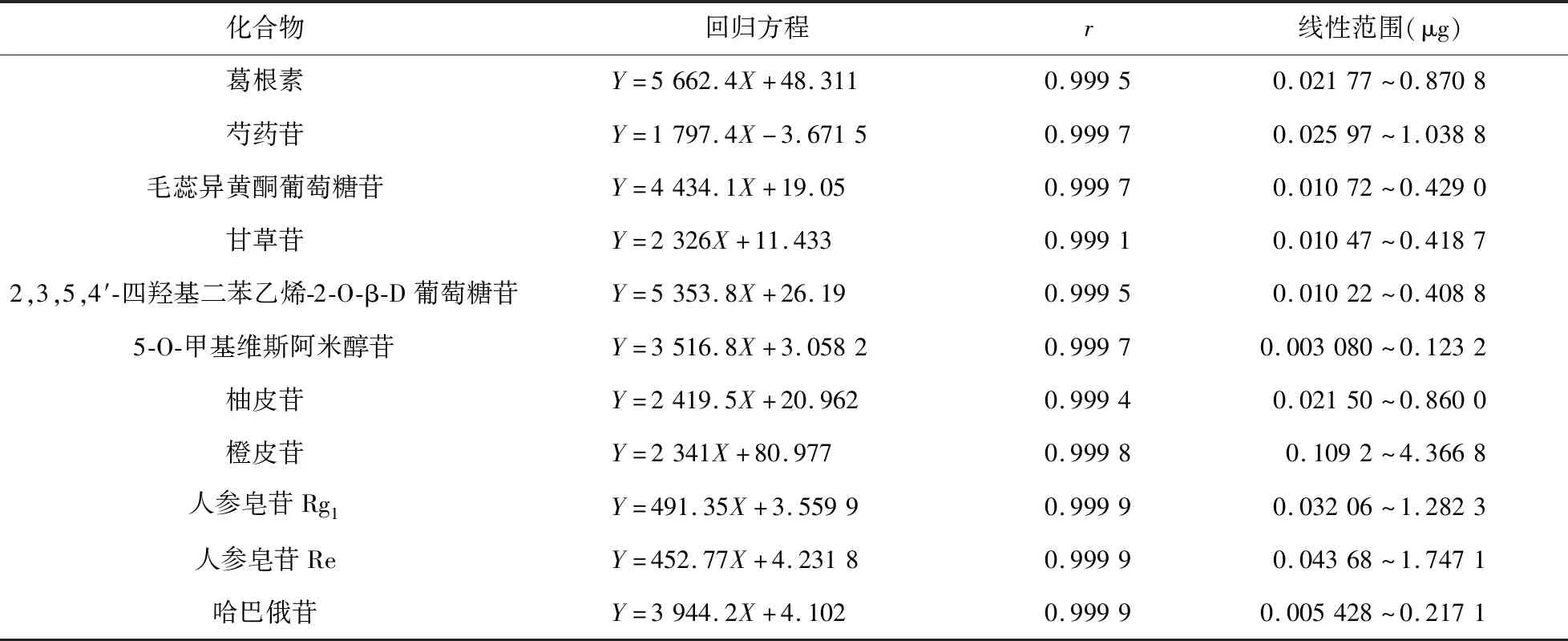
表1 11个成分的回归方程、相关系数及线性范围
2.4 专属性 将样品中与对照品保留时间相对应的色谱峰分别与各个对照品色谱峰在190~400 nm做光谱匹配,样品与对照品DAD光谱匹配度均大于990。阴性样品对测定无干扰。
2.5 精密度试验 精密吸取标准曲线第2点的对照品混合溶液,连续进样6次,计算葛根素、芍药苷、毛蕊异黄酮葡萄糖苷、甘草苷、2,3,5,4′-四羟基二苯乙烯-2-O-β-D葡萄糖苷、5-O-甲基维斯阿米醇苷、柚皮苷、橙皮苷、人参皂苷Rg1、人参皂苷Re、哈巴俄苷峰面积的RSD,分别为1.1%、0.9%、0.6%、0.7%、1.5%、1.2%、0.6%、1.0%、0.9%、1.1%、0.8%,表明精密度良好。
2.6 稳定性试验 取同一供试品溶液,按“2.2”项下色谱条件分别在0、4、8、12、16、24 h依法进样测定,测得供试品溶液中葛根素、芍药苷、毛蕊异黄酮葡萄糖苷、甘草苷、2,3,5,4′-四羟基二苯乙烯-2-O-β-D葡萄糖苷、5-O-甲基维斯阿米醇苷、柚皮苷、橙皮苷、人参皂苷Rg1、人参皂苷Re、哈巴俄苷峰面积的RSD分别为0.8%、1.0%、1.8%、0.9%、2.0%、1.8%、0.7%、1.3%、1.3%、1.5%、1.1%,表明供试品溶液在24 h内稳定。
2.7 重复性试验 取同一批号(QB2027)人参再造丸,按“2.1.2”项下方法制备供试品溶液,按“2.2”项色谱条件测定,进行6次平行试验,结果葛根素、芍药苷、毛蕊异黄酮葡萄糖苷、甘草苷、2,3,5,4′-四羟基二苯乙烯-2-O-β-D葡萄糖苷、5-O-甲基维斯阿米醇苷、柚皮苷、橙皮苷、人参皂苷Rg1、人参皂苷Re、哈巴俄苷的含量平均值分别为0.21、0.27、0.052、0.085、0.20、0.013、0.093、1.4、0.11、0.21、0.011 mg/g,RSD依次为2.8%、2.3%、1.5%、2.2%、3.0%、2.9%、1.8%、2.8%、2.7%、2.6%、2.7%,表明本方法重复性良好。
2.8 加样回收率试验 取已知含量(批号:QB2027)的人参再造丸样品9份,每份1 g,精密称定,分为3组,每组3份,分别精密加入含葛根素232.78 μg/ml、芍药苷268.57 μg/ml、毛蕊异黄酮葡萄糖苷51.02 μg/ml、甘草苷82.37 μg/ml、2,3,5,4′-四羟基二苯乙烯-2-O-β-D葡萄糖苷191.28 μg/ml、5-O-甲基维斯阿米醇苷13.28 μg/ml、柚皮苷99.11 μg/ml、橙皮苷1 431.89 μg/ml、人参皂苷Rg1105.73 μg/ml、人参皂苷Re 202.32 μg/ml、哈巴俄苷10.25 μg/ml的混合对照品溶液0.5、1.0、1.5 ml,至氮吹仪上吹干,按照“2.1.2”项下方法制备供试品溶液,依法测定,计算11种成分的回收率,如表2所示。结果表明,2,3,5,4′-四羟基二苯乙烯-2-O-β-D葡萄糖苷不稳定,平均回收率为87.1%,其余10个成分平均回收率在90%~100%之间,RSD均小于5.0%,满足《中国药典》2015年版四部通则9101“药品质量标准分析方法验证指导原则”的要求。
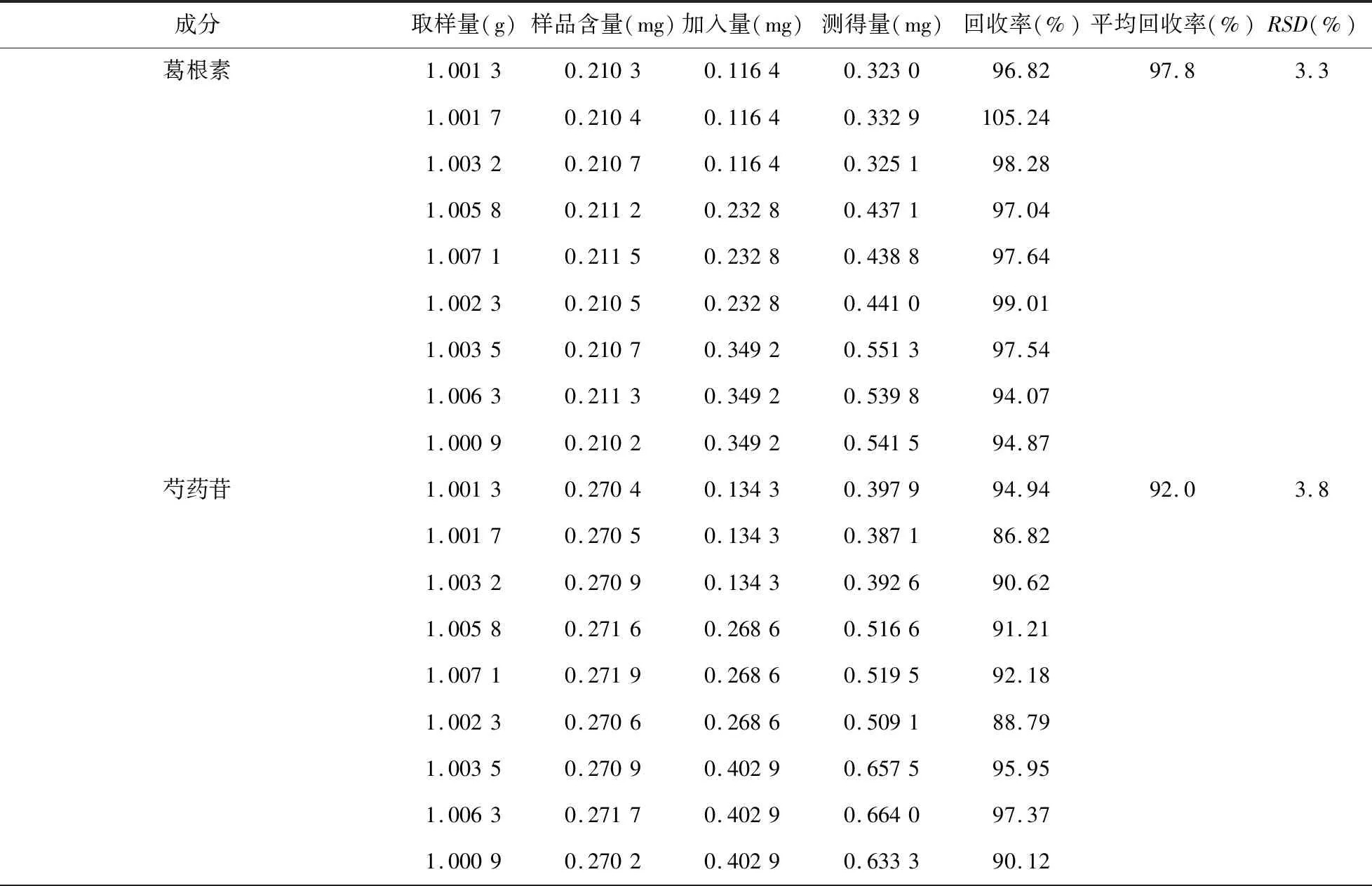
表2 11个成分的加样回收率(n=9)
2.9 样品测定 取不同厂家的各批样品,分别按“2.1.2”项下的方法制备供试品溶液,按“2.2”项色谱条件进行分析,记录峰面积,按外标法计算。结果见表3。
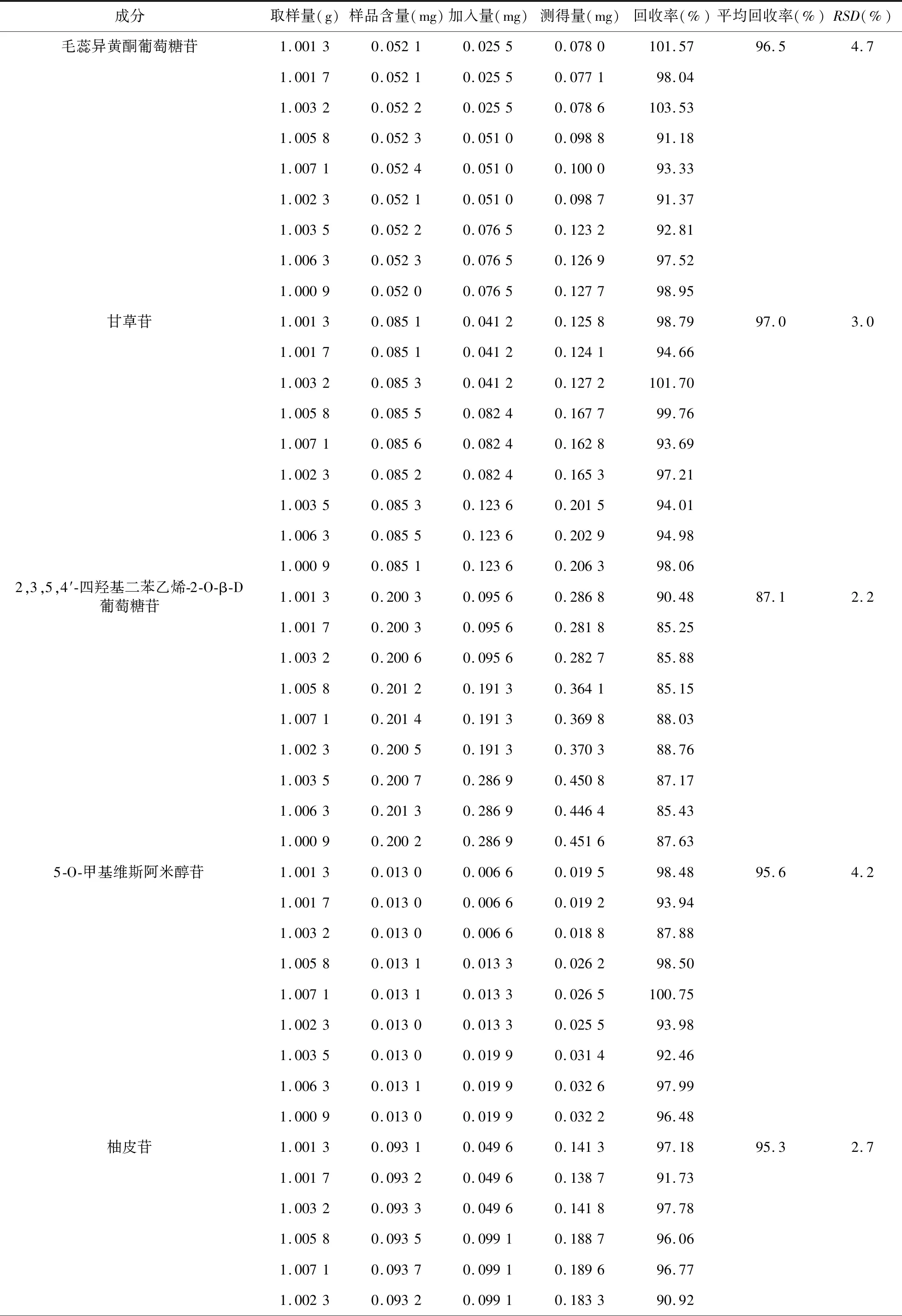
续表2
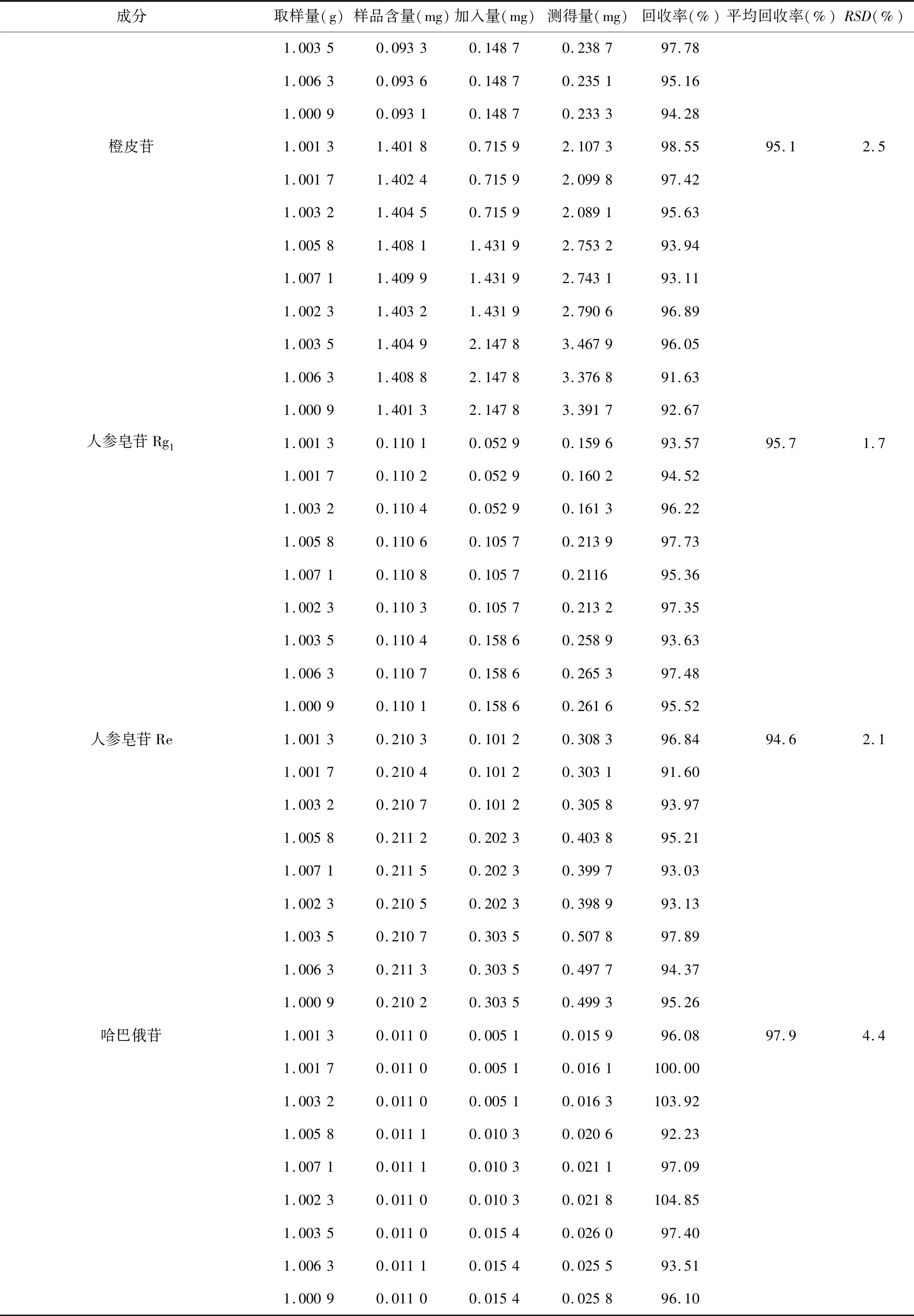
续表2
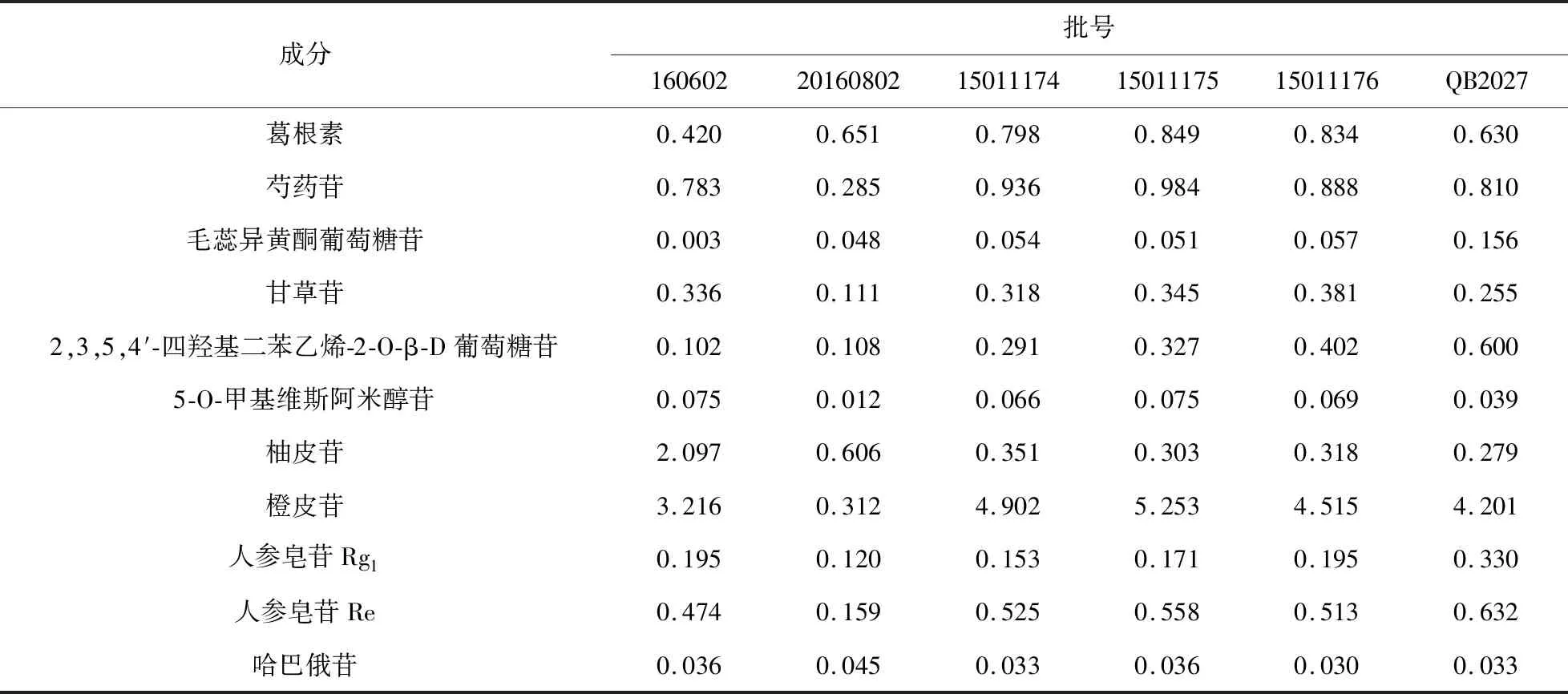
表3 样品含量测定结果(mg/丸,n=3)
3 讨论
3.1 提取条件的选择 由于人参再造丸处方组成很多,化学成分复杂,各化合物结构类型丰富,理化性质差异较大,提取分离比较困难,同一种提前分离方法难以满足很多化合物的测定,本研究经过大量的试验摸索,筛选测定了上述11种化合物。
本试验分别选取了超声、回流、索氏提取3种提取方法,选用了乙醚、三氯甲烷、石油醚(60~90 ℃)3种除杂方法,选择了中性氧化铝柱、D101型大孔吸附树脂柱、聚酰胺柱3种柱层析方法来纯化,结果表明,选用索氏提取、使用石油醚(60~90 ℃)除杂、经D101型大孔吸附树脂柱纯化的方法能使待测成分提取完全,且杂质较少。其中,D101型大孔吸附树脂柱的预处理、吸附与解吸对加样回收率影响较大,是提取过程的重点。本试验同时考察了加入甲醇后的回流时间,对比了1 h、3 h、5 h、7 h的结果,大于3 h后2,3,5,4′-四羟基二苯乙烯-2-O-β-D葡萄糖苷的含量下降,综合分析考虑选择回流时间为3 h。
由于大蜜丸在溶剂中很难散开,需要剪碎后加硅藻土来加快混匀溶散并辅助指标性成分的滤出,本试验考察了样品和硅藻土的比例,发现等比例混匀条件下提取效果最好。
3.2 色谱条件的选择 本试验同时测定多种化合物,对色谱条件要求较高,流动相尝试了甲醇、乙腈、水、磷酸、冰醋酸多种比例的组合,结果发现,乙腈-0.05%磷酸溶液系统分离效果好、干扰小、峰形好。同时,因为本药处方组成很多,每一味药材比例较低,因此比较适合选用各成分的最大吸收波长来测定,本试验利用DAD光谱扫描,结合各色谱峰强度和干扰情况,选择了较为理想的不同波长。
3.3 结果分析 4个生产厂家的人参再造丸中各成分含量差异较大,其中橙皮苷最大,可相差近17倍,可能因不同厂家的工艺不同、投料差异等原因引起。影响中药材成分含量的因素较多,包括土壤、光照、环境温度、降水量、农艺措施、采收时间、药材初加工、贮运方式、药材掺杂造假等[14],基于中成药成分的复杂性,中成药的质量控制应该从多指标检测进行综合评价。
4 结论
本研究建立了HPLC-DAD法同时测定人参再造丸中11个成分含量的方法,覆盖了方中的多味药材,是现行药典标准只测定盐酸小檗碱的有力补充,该方法分离度良好,测定结果稳定、重复性好,能够为高效、准确地控制人参再造丸的质量提供新思路。
猜你喜欢
食品安全导刊(2020年30期)2020-11-23
医学新知(2019年4期)2020-01-02
中国食品(2018年7期)2018-09-10
幸福·健康版(2018年3期)2018-03-23
食品与健康(2017年9期)2017-09-13
家庭医药·快乐养生(2017年4期)2017-04-19
恋爱婚姻家庭·养生版(2017年2期)2017-02-15
药学研究(2015年11期)2015-12-19
饮食科学(2015年2期)2015-09-24
癌变·畸变·突变(2015年3期)2015-02-27
