
猪流行性腹泻病毒蛋白拮抗宿主天然免疫应答的研究进展
2020-12-21 09:20成温玉白云贾怀杰强桃艳赵鸿远张博艺郭晓荟
生物技术通报 2020年12期
成温玉 白云 贾怀杰 强桃艳 赵鸿远 张博艺 郭晓荟
(1. 安康学院 现代农业与生物科技学院,安康 725000;2. 中国农业科学院兰州兽医研究所 家畜疫病病原生物学国家重点实验室农业农村部兽医公共卫生重点实验室,兰州 730046)
腹泻病是困扰当前养猪业的主要疾病之一,不仅影响猪群的生产性能、降低饲料转化率和增加养殖成本,而且还会引起猪只的死亡,干扰猪肉市场的稳定性。冠状病毒是引起猪腹泻病主要病毒性病原之一,其中以猪流行性腹泻病毒(Porcine epidemic diarrhea virus,PEDV)引起的腹泻病尤为严重,可导致仔猪100%的发病率和死亡率,患畜主要表现为厌食、呕吐、严重腹泻和脱水等症状[1]。自1978年首次在英国发现PED以来,全球各大养猪国家均有报道且均发生过爆发的情况,是当前养猪业重视的猪病之一[2-3]。在我国,早在1984年就确认并分离到PEDV,而在随后的26年里该病毒仅引起部分猪场零星发病,直到2010年10月PEDV引起的猪急性腹泻突然爆发并迅速席卷全国,给养猪业造成严重的经济损失[2-3]。
天然免疫是机体非特异性抵御病原感染和有害物质入侵的第一道防线,并启动和参与获得性免疫应答。天然免疫系统中的多种成分包括模式识别受体、信号转导及调节因子、Ⅰ型-III型干扰素(Interferon,IFN)、干扰素刺激基因(Interferonstimulated genes,ISGs)和趋化因子从多个层面参与并抑制病原的感染。然而,病原在与宿主长期的相互作用过程中进化出一套能够逃逸和破坏宿主免疫系统的机制,通过拮抗宿主天然免疫信号通路的激活,逃逸免疫应答,促进自身增殖,维持其在宿主体内的生存[4]。PEDV具有典型的免疫抑制能力,依赖于自身编码的免疫拮抗蛋白和隐藏自身病原体相关分子模式(dsRNA)逃逸宿主的免疫反应[5]。研究证实,PEDV至少编码10种蛋白参与拮抗宿主天然免疫反应,但目前人们对其逃逸机制认识不深[6]。为了进一步认识PEDV拮抗宿主天然免疫应答机制,深入探究病原的致病机理,本文就PEDV蛋白如何拮抗宿主天然免疫应答进行总结,以期为疫苗的创制奠定基础。
1 PEDV基因组结构
与其他冠状病毒科的成员类似,PEDV属于囊膜病毒,包含单股正链RNA基因组,由至少7个开放阅读框(Open reading frame,ORF)以5'UTRORF1a-ORF1b-S-ORF3-E-M-N-3'UTR为顺序组成近乎28 nt大小的基因组,共编码2个多聚蛋白前体(pp1a和pp1ab)、4个结构蛋白(S、E、M和N)和1个辅助蛋白(ORF3)。其中基因组5'端的ORF1a和ORF1b基因占据整个基因组2/3,编码的两个病毒复制酶多聚蛋白(pp1a和pp1ab)经剪切加工后形成16个成熟的非结构蛋白(nsp1-nsp16),参与病毒RNA的复制和转录;其余靠近3'端的1/3基因组编码4个结构蛋白和辅助蛋白ORF3[7]。值得注意的是,编码辅助蛋白ORF3的基因位于S基因和E基因之间,属PEDV特异性基因,与病毒毒力密切相关[8]。
2 PEDV蛋白抑制宿主天然免疫应答
2.1 非结构蛋白
冠状病毒非结构蛋白属于早期蛋白,主要参与病毒RNA的合成。然而越来越多的研究发现,其中nsp1、nsp3、nsp5、nsp7、nsp14、nsp15和nsp16还参与对宿主免疫调节的功能[5-6,8]。在病原感染早期,上述非结构蛋白通过多种方式抑制宿主天然免疫应答,为病毒的侵入和复制创造机会。
2.1.1 nsp1 nsp1大小为110个氨基酸,是pp1a蛋白N端剪切后的产物,仅表达于α和β冠状病毒属的各成员中,但其序列在α和β属各成员中却表现出较大的变异性,是种属特异性基因[9]。PEDV nsp1定位在宿主细胞的线粒体、内质网和高尔基体中,参与病毒基因的表达[10]。近几年,多个研究发现nsp1也参与了宿主基因调节并逃避宿主免疫应答(图1)。在拮抗宿主干扰素通路中,nsp1依赖于蛋白酶体途径降解宿主胞核中的CBP蛋白,使得后者不能与IRF3组装,从而阻碍I型干扰素及其ISGs的表达[11]。同时nsp1也能够抑制III型干扰素IFN-λ的抗病毒活性[12]。研究发现,在PECDQ,LLC-PK1和MARC-145细胞中,nsp1通过阻碍IRF1入核,降低细胞中病毒活化的过氧化物酶体数量来干扰IFN-λ的产生。进一步利用突变体的研究发现,nsp1干扰IFN-λ产生的能力主要依赖于其序列中保守的氨基酸区域[12]。同时,对同属α冠状病毒TGEV nsp1的结构研究证实,第91-95位氨基酸序列高度保守,是构成nsp1蛋白抑制宿主调控免疫应答的主要基序,决定TGEV的毒力[13]。
除拮抗宿主干扰素通路外,nsp1还具有阻碍NF-κB核转移,影响IFN-β及多个细胞因子(TNF-α、IL-1β、IL-6、IL-15和IL-17)表达的能力[14]。在PEDV感染LLC-PK1细胞早期,促炎性细胞因子的表达受到抑制,nsp1通过靶向干扰IκBα的磷酸化和泛素化,阻碍了p65亚基的核移位,从而切断NF-κB信号激活通路,抑制一系列抗病毒细胞因子的表达(图1)。
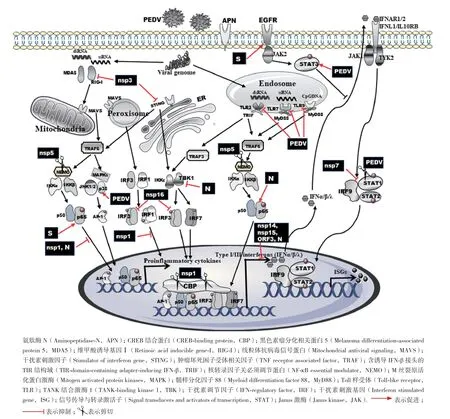
图1 PEDV蛋白拮抗宿主天然免疫信号通路
2.1.2 nsp3 冠状病毒nsp3是一个多结构域的大分子蛋白,包含重要的泛素样结构域和木瓜样蛋白酶(Papain-like protease,PLpro)结构域,且这些结构域在不同种属的冠状病毒成员中存在较大的结构差异。nsp3因其具有PLpro活性,在病毒复制过程中,参与了病毒复制转录复合体的形成,依赖于自身PLpro结构域与nsp5的3CLpro结构域共同完成了对前体多聚蛋白pp1a和pp1ab的加工[15]。同样,nsp3拮抗宿主天然免疫应答也依赖于其PLpro活性。Xing等[16]研究证实,PEDV nsp3蛋白借助PLpro的去泛素化活性阻碍STING和RIG-I泛素化,从而干扰STING和RIG-I介导的I型干扰素通路的激活,致使宿主I型干扰素以及ISGs的表达受阻(图1)。此外,来自于对SARS-CoV和TGEV nsp3的研究证实,nsp3还可通过抑制IκBα的泛素化以及p56的磷酸化阻止NF-κB介导的细胞因子应答反应[17]。因此,nsp3抗宿主免疫应答主要依赖于其PLpro活性,而nsp3是否具有其他抑制宿主免疫应答的机制有待进一步探究。
2.1.3 nsp5 nsp5是一种具有蛋白水解功能的3C样蛋白酶(3C-like protease,3CLpro),富含组氨酸和半胱氨酸,在所有冠状病毒间具有很高的保守型。该蛋白是冠状病毒的主蛋白酶,通常以二聚体的形式负责对病毒多聚前体蛋白pp1ab进行切割形成nsp4-nsp16[18]。除具有切割病毒自身蛋白的功能外,nsp5也能切割宿主蛋白。Wang等[19]起初发现,nsp5具有降低仙台病毒(Sendai virus,SEV)诱导IFN-β表达能力的现象,进一步研究证实nsp5能够靶向切割天然免疫RIG-I/MDA5信号通路上的重要接头分子NEMO,从而破坏宿主产生I型干扰素。nsp5依赖于其保守的组氨酸和半胱氨酸催化位点切割NEMO位于第231位的谷氨酸残基,致使NEMO丧失激活下游信号分子的能力,阻碍I型干扰素生成(图1)。同样,PDCoV的nsp5也能通过切割宿主NEMO抑制IFN-β的产生,且切割位点也位于NEMO的第231氨基酸残基,这与nsp5在冠状病毒中具有较高保守性密切相关[20]。此外,PDCoV的nsp5还能抑制IFN-α介导的ISGs转录,其通过靶向切割I型干扰素下游JAK-STAT通路中的关键分子STAT2抑制ISGs的表达,从而拮抗I型干扰素的信号转导[21]。尽管nsp5切割STAT2的能力仅存在于PDCoV中,但有研究发现PEDV能够抑制感染细胞中STAT1的表达。Guo等[22]发现,PEDV依赖于宿主的泛素蛋白酶系统降解病毒诱导的STAT1,进而降低其下游I型干扰素的表达(图1)。另外,在PEDV感染的细胞中出现STAT3被激活的现象,作为负调控I型干扰素表达的JAK2-STAT3通路中关键分子STAT3,其上游的关键性配体——表皮生长因子(Epidermal growth factor,EGF)在被PEDV激活的EGFR竞争性结合后处于激活状态,导致I型干扰素表达受阻[23](图1)。上述PEDV通过降解STAT1和激活STAT3途径抑制干扰素表达的能力已经得到充分证实,但参与调节此过程的病毒蛋白尚未明确,有待进一步验证。
2.1.4 nsp7 PEDV非结构蛋白7是一包含83个氨基酸的蛋白质,在不同毒株间高度保守,主要定位于细胞质[24]。病毒在复制过程,nsp7与nsp8形成多聚体复合物,参与病毒RNA合成和病毒粒子组装。同时,多篇文献证实nsp7具有调节宿主I型干扰素表达的功能[11,24-25]。利用双荧光素酶报告系统和荧光定量PCR证实,nsp7能够抑制IFN-α介导的ISRE启动子活化和ISGs的表达,进一步研究发现PEDV nsp7通过与宿主转运蛋白α1(Karyopherin α1,KPNA1)竞争性结合STAT1,阻碍了KPNA1与STAT1的相互识别,导致干扰素刺激因子3(Interferon-stimulated gene factor 3,ISGF3)核转运受阻,从而抑制I型干扰素及其相关基因的表达[25]。ISGF3是STAT1、STAT2与IRF9三者形成的复合体,nsp7除了与STAT1结合之外,还可以靶向结合于STAT2的DBD结构域(DNA-binding domain),但并不影响ISGF3的形成[25](图1)。
2.1.5 nsp15 nsp15是冠状病毒的内切核糖核酸酶,参与病毒RNA的合成。起初,nsp15被认为是病毒复制复合体的重要组分,后来的研究发现其并非在病毒复制时所必须,而是在拮抗宿主免疫应答中发挥重要作用[26]。Zhang等[11-12]通过双荧光素报告酶系统筛选抑制干扰素应答功能的PEDV蛋白质时,鉴定出nsp15分别具有抑制Poly(I:C)诱导的IFN-β和IFN-λ激活的功能。Deng等[27]通过反向遗传技术建立的PEDV感染性克隆证实,nsp15必须依赖其内切核糖核酸酶活性拮抗宿主I型(IFN-β)和III型干扰素(IFN-λ)应答。病毒丧失核糖核酸酶活性后激活细胞(LLC-PK1)中I型(IFN-β)和III型干扰素大量表达,导致其在猪上皮细胞中的增殖能力减弱。同时,丧失核糖核酸酶活性的PEDV对仔猪的致病力也显著降低[27]。然而,针对PDCoV nsp15的研究发现,该蛋白也具有抑制SEV诱导的IFN-β激活的能力,但并非依赖于nsp15核糖核酸酶活性,而是通过靶向抑制NF-κB p65亚基的激活和核移位阻断下游I干扰素通路的活化,从而拮抗宿主的免疫应答[28]。上述研究结果表明,PEDV和PDCoV依赖于nsp15通过不同形式拮抗宿主天然免疫应答,然而我们依然不明确PEDV如何利用nsp15的核糖核酸酶活性发挥抗宿主I型(IFN-β)和III型干扰素(IFN-λ)应答。有文献推测,病毒在复制时nsp15与其他非结构蛋白在宿主细胞中构成双膜结构的小囊泡,包裹病毒RNA以逃避其被宿主核酸识别受体的识别[28],但其具体机制有待进一步证实。
2.1.6 nsp16 冠状病毒编码的nsp16是一种2'氧位甲基转移酶,主要参与病毒RNA的合成,其氨基酸序列高度保守,存在Lys-Asp-Lys-Glu(KDKE)四个一组的氨基酸基序是维持甲基转移酶活性的关键[29]。dsRNA作为RNA病毒复制过程中的中间产物,其能被宿主模式识别受体(PPRs)识别并诱导机体干扰素应答。为了逃避宿主免疫系统对dsRNA的识别,冠状病毒在复制过程中,借助nsp14的甲基转移酶结构域和nsp16甲基转移酶活性与nsp10协同作用对病毒RNA的5'端进行加帽反应,避免病毒RNA受宿主核酸识别受体的识别[29]。针对PEDV拮抗宿主干扰素的研究发现,nsp16依赖其甲基转移酶活性通过抑制IRF3磷酸化阻断RIG-I和MDA5引发的天然免疫信号通路发挥抗宿主免疫反应,其保守的KDKE基序是阻碍IFN-β和ISRE启动子激活的关键[30](图1)。当人为缺失或者突变病毒nsp16的KDKE基序后,病毒诱发强大的干扰素应答反应[30]。同时nsp10具有协同nsp16抑制宿主I型干扰素应答的能力,而nsp10自身却无此功能,这可能与nsp10参与激活nsp16甲基转移酶活性有关[31]。同样,来自MERS-CoV nsp16的研究也证实其具有拮抗宿主I型干扰素应答的能力,并且突变其保守KDKE基序的病毒对小鼠具有很好的免疫保护作用[32]。因此,nsp16可作为疫苗设计的良好靶点。
除上述几种非结构蛋白外,nsp14也被证实能够抑制IFN-β启动子活性,nsp8和nsp14具有抑制III型干扰素IFN-λ1启动子活性的能力[11-12],但其具体的作用机制尚未见报道。我们对nsp14的功能预测显示,其具有外切酶核酸活性和N7甲基转移酶活性。利用反向重组技术构建的缺失N7甲基转移酶活性的PEDV感染细胞后,病毒的增殖不受影响但对外源性IFN-β敏感性增加,表明nsp14的N7甲基转移酶活性在抗IFN-β的应答中起到关键性的作用(待发表)。然而目前我们尚未发现nsp14拮抗宿主I型干扰素应答具体机制。
2.2 结构蛋白
在位于PEDV基因组的3'端,编码4个结构蛋白,即棘突蛋白S(Spike)、包膜蛋白E(Envelope)、膜蛋白M(Membrane)和核衣壳蛋白N(Nucleocapsid),主要参与病毒构成。利用异位表达研究发现,其中E、N和M表现出抑制IFN-β和IRF3活性的能力。此外还参与宿主内质网应激和NF-κB激活反应。
2.2.1 棘突蛋白S 棘突蛋白S属于I型糖蛋白,包含S1和S2两个亚基并以三聚体的形式存在于病毒粒子表面,决定病毒的宿主范围和组织嗜性[33]。S1亚基又含有N端区域(N-terminal domain,S1-NTD)和 C端 区 域(C-terminal domain,S1-CTD),二 者均属于PEDV的受体结合域,其中S1-CTD识别并特异性结合宿主受体氨肽酶N(Aminopeptidase N,APN),而S1-NTD则可以与病毒的辅助性受体糖结合[34]。病毒入侵细胞时借助于S蛋白与宿主细胞表面受体结合并通过膜融合的形式进入胞内。尽管利用双荧光素酶报告系统未发现S蛋白具有抑制I型干扰素的能力,然而在PEDV感染的肠细胞中,S蛋白表现出削弱宿主I型干扰素应答的潜能[23]。Yang等[23]发现,S蛋白能与宿主表皮生长因子受体(Epidermal growth factor receptor,EGFR)结 合并使EGFR激活,活化后的EGFR进一步激活下游JAK2-STAT3通路。作为负调控I型干扰素表达的STAT3被激活后,反过来抑制I型干扰素的产生,从而促进病毒感染。此外,S1亚基具有诱导细胞凋亡的能力[35],虽然细胞凋亡阻碍了病毒复制并暴露细胞中的病毒,但同时也有利于病毒释放以及逃避宿主胞内强烈的免疫应答反应。
2.2.2 囊膜蛋白E和膜蛋白M E蛋白是由76个氨基酸组成的分子量约为 8.8 kD最小的结构蛋白,在PEDV出芽过程中扮演重要的角色[36]。该蛋白在病毒感染过程中主要定位于内质网,有研究证实其能够通过上调宿主葡萄糖调节蛋白(Glucose-regulated protein 78,GRP78)的表达诱导感染细胞的内质网应激反应,同时激活NF-κB活性并上调炎症因子IL-8和抗凋亡分子Bcl-2的表达[37]。据此,该研究推测E蛋白通过激活NF-κB上调IL-8和Bcl-2的表达分别促进细胞炎性反应和限制细胞凋亡,维持病毒在细胞中增殖。
膜蛋白M分子量大小约为30 kD,在冠状病毒中高度保守,定位于整个宿主细胞中,主要参与病毒粒子的组装和出芽。早期研究发现,PEDV M蛋白可诱导宿主产生IFN-α和特异性抗体,还能介导细胞融合,因此被作为病毒抗体疫苗的候选靶点[38]。与E蛋白类似,尽管M蛋白不具有拮抗I型和III型干扰素的能力,然而其能够抑制细胞周期素A(cyclin A)阻碍肠道上皮细胞生长使其停滞在细胞周期的S期,暗示病毒可能通过干扰细胞周期促进其增殖[38]。
2.2.3 核衣壳蛋白N N蛋白是所有PEDV编码的结构蛋白中表达最丰富、最保守的蛋白,大小为55-58 kD,主要定位于宿主细胞胞质中,参与病毒基因组转录和合成、病毒粒子组装以及宿主应激和免疫反应[39]。N蛋白作为病毒逃逸宿主免疫应答策略的一部分,具有拮抗I型和III型干扰素的能力。利用双荧光素酶报告实验显示,异位表达的N蛋白分别具有抑制SEV诱导的IFN-β产生和poly(I:C)诱导的IFN-λ3的启动子活性,同时还能抑制转录因子IRF3和NF-κB的激活[11-12](图1)。作为RIGI/MDA5介导I型干扰素通路中的关键蛋白,TBK1受到N蛋白的靶向作用,从而阻断了TBK1对IRF3的激活,造成下游IFN-β和ISGs表达受阻[40]。在抑制IFN-λ3表达的通路中,N蛋白被发现能够阻止IPEC-J2细胞中NF-κB和IRF3的激活,因此N蛋白有可能通过抑制NF-κB和IRF3的激活破坏IFN-λ3对病毒的应答[41]。同样,在Zhang等[12]对PEDV蛋白抑制III型干扰素表达的筛选研究中,也发现N蛋白具有拮抗IFN-λ1活性的能力,但其具体机制尚未深入研究。
然而,与上述N蛋白抑制NF-κB激活的现象相反,Cao等[42]发现在IECs细胞中过表达PEDV的N蛋白能显著激活NF-κB,且NF-κB的激活活性依赖于N蛋白的表达剂量和蛋白的中间序列,N蛋白对NF-κB的激活能力主要依靠宿主TLR2信号通路的参与(图1)。同时他们还发现TLR3和TLR9也参与了PEDV诱导的NF-κB激活,但其具体机制未见报道。此外,Xu等[43]也证实N蛋白能激活IECs细胞中NF-κB并诱导细胞内质网应激反应、上调IL-8和Bcl-2的表达,同时还能通过降解细胞周期素A阻碍肠道上皮细胞生长使其停滞在细胞周期的S期。这与E蛋白的作用相似,很可能N蛋白也是通过限制细胞凋亡,利于病毒在细胞中复制。另外发现,N蛋白可穿梭于胞质和胞核之间,与核仁磷酸蛋白1(Nucleophosmin,NPM1)存在相互作用[44]。NPM1作为凋亡抑制蛋白,受caspase-3的靶向切割后会促进细胞凋亡。在PEDV感染的过程中,NPM1表达上调且过表达能促进PEDV的增殖,尽管N蛋白入核过程不依赖于NPM1的作用,但N蛋白的结合保护NPM1免受caspase-3的水解,利于细胞生存,促进病原的增殖[44]。
2.3 辅助蛋白
ORF3作为PEDV唯一的辅助蛋白,大小约为25 kD,与病毒的毒力强弱和复制密切相关。对比野生、细胞培养株和弱毒疫苗株的ORF3序列发现,弱毒疫苗株和细胞培养株出现30-51核苷酸的缺失,因此常被用于强弱毒株诊断的依据[45]。ORF3主要定位于细胞质中,部分存在于内质网和高尔基体等细胞器,其可与S蛋白共定位于感染细胞中的核周间隙和囊泡状结构中,且二者存在相互作用调节病毒复制[46]。尽管通过双荧光素酶报告实验筛选到ORF3也具有抑制I型(IFN-β)和III型(IFN-λ1)干扰素启动子活性的能力[11-12](图1),但对其抑制IFN-β和IFN-λ1的具体机制及作用效果并未进行深入研究。与结构蛋白M和N相似,ORF3也表现出调控宿主细胞周期的能力。Ye等[47]利用构建稳定表达ORF3蛋白的Vero细胞证实,ORF3通过延长细胞周期的S期和促进囊泡形成的方式以利于病毒增殖。同时相比强毒株的PEDV,稳定表达ORF3的Vero细胞更利于弱毒株的增殖。此外,ORF3还具有抑制PEDV诱导的细胞凋亡功能,利用反向遗传构建的PEDV-ΔORF3感染细胞后,细胞活性显著降低且活化的caspase-3升高,表明缺失ORF3的病毒促进了细胞凋亡[48]。因此,ORF3在病毒感染过程中不仅可以通过抑制I型和III型干扰素的表达拮抗宿主天然免疫应答,而且能够直接调控细胞周期、抑制细胞凋亡为病毒增殖创造细胞环境。
3 PEDV拮抗宿主免疫的其他方式
PEDV除了利用上述蛋白拮抗宿主免疫应答外,还采用其他方式逃逸宿主的免疫反应。丝裂原活化蛋白激酶(Mitogen-activated protein kinase,MAPK)通路是机体调控胞内外信号网络的关键,其通路中的关键元件包括胞外信号调节激酶(Extracellular signal-regulated kinases,EKR)、c-Jun氨基末端激酶(c-Jun N-terminal kinases,JNK)和p38。研究发现,PEDV在感染过程中能激活MAPK通路中的EKR和p38促进病毒复制,而被激活的EKR和p38并不能诱导宿主细胞周期停滞和凋亡反应[49](图1)。鉴于PEDV和ORF3蛋白均具有抑制细胞凋亡的作用,因此,尽管PEDV能激活EKR和p38但不诱发细胞凋亡反应,这是病毒调控宿主反应的综合结果,而PEDV是如何激活EKR和p38并促进自身增殖有待进一步探究。
除了RIG-I和MDA5受体及其信号通路受到PEDV调控外,TLRs家族中的TLR3、TLR4和TLR7-TLR9及其下游关键分子TRIF、MyD88和TRAF6在病毒感染仔猪早期时(4 dpi)表达受到抑制,进而阻碍新生儿Fc受体(Neonatal Fc receptor,FcRn)表达及IgG的分泌[50]。同样,在对比S基因变异毒株(S-INDEL strain)和非变异毒株(non-SINDEL strain)对仔猪免疫应答的影响研究中发现,仔猪肠黏膜中TLR3、TLR4、TLR7-TLR9、TRIF、MyD88和TRAF6的表达也明显受到抑制,且这些基因的表达在非变异毒株感染的仔猪中被下调的幅度最大,暗示S蛋白在抑制宿主TLRs信号通路中发挥作用[51]。HSP27是参与IFN-β及其下游ISGs表达的重要分子,通过激活NF-κB促进细胞中IFN-β及其下游ISGs转录,具有抗病毒感染的作用。PEDV在Marc-145细胞中增殖过程表现出对HSP27的抑制功能,从而降低IFN-β及其下游ISGs的表达,逃避宿主抗病毒应答[52]。
4 总结与展望
病毒与宿主之间的相互作用像是二者之间的一场“军备竞赛”,病毒为了生存依赖于自身编码的蛋白隐藏病原相关分子模式(PAMPs)逃避并直接阻碍宿主的免疫应答。猪细胞中的TLRs和RLRs能够识别PEDV在增殖过程中释放的基因组RNA以及形成的核酸中间物并激活天然免疫抗病毒途径,产生I型、III型干扰素和大量的ISGs。然而,PEDV编码的10余种蛋白能够通过两种方式拮抗宿主干扰素应答:(1)靶向作用于干扰素通路中的多个信号分子阻断I型和III型干扰素产生,甚至直接抑制干扰素下游ISGs的生成;(2)借助于病毒蛋白的酶活性修饰RNA结构,逃避宿主对其PAMPs的识别。此外,病毒还通过调控宿主细胞以延长细胞周期或阻碍感染细胞的凋亡为自身增殖创造细胞环境,而随着细胞中病毒粒子增加以及相关凋亡信号的累积,细胞不得不启动凋亡时,又为病毒的释放和扩散创造了条件。因此,PEDV不仅通过阻碍免疫应答拮抗宿主的清除,还可调控宿主细胞周期和细胞凋亡反应方便自身增殖。
尽管PEDV基因组结构相对简单,除了已知参与调控宿主免疫应答的蛋白之外,其他非结构蛋白、结构蛋白以及基因组两端UTR序列是否具有拮抗和调控宿主天然免疫应答的潜力;nsp8和nsp14拮抗I型和III型干扰素应答的具体机制是什么;它们对病毒的毒力有何影响;病毒诱导的细胞凋亡与天然免疫应答以及凋亡抑制之间有何关系;PEDV感染引起宿主细胞的内质网应激反应和自噬的机制是什么。因此,从宿主免疫学的角度出发,探究病毒编码蛋白对宿主的免疫调节作用有助于认识病毒与宿主相互作用关系,同时对疫苗的创制和疾病防控具有重要的指导意义。
猜你喜欢
广西医学(2022年22期)2023-01-30
科学(2020年3期)2020-11-26
当代水产(2020年3期)2020-06-15
中国继续医学教育(2015年6期)2016-01-07
中国病理生理杂志(2015年8期)2015-12-21
小星星·阅读100分(高年级)(2015年11期)2015-11-28
中国医疗美容(2015年2期)2015-07-19
中国生化药物杂志(2015年4期)2015-07-07
医学研究杂志(2015年12期)2015-06-10
医学研究杂志(2015年3期)2015-06-10
