
微生物宏基因组数据分析方法研究进展
2020-12-23 10:57赵圣国王加启
微生物学杂志 2020年5期
李 敏, 赵圣国, 郑 楠, 王加启*
(1中国农业科学院湖北北京畜牧兽医研究所 动物营养学国家重点实验室,北京 100193;2.华中农业大学动物科技学院 动物医学院,湖北 武汉 430000)
微生物主要包括细菌、真菌、古生菌和原虫等,是生态系统中不可缺少的一部分。微生物不论对人类还是动植物的营养健康、行为活动都起着重要的作用。近年来,肠道微生物与宿主之间的关系成为研究热点。人体肠道微生物可以影响人的生长发育和健康疾病,对人们的生活至关重要[1]。通过对动物肠道微生物的研究发现,反刍动物微生物在维持宿主的能量代谢、营养吸收和生理性能等方面发挥着重要作用[2]。 微生物宏基因组是指全部微生物遗传物质总和,它包含了可培养微生物和不可培养微生物基因,最早是在1998年由Handelsman等[3]研究土壤微生物提出的,通过宏基因组的克隆获得了土壤微生物群落的全部基因集,数据分析表明土壤宏基因组中所包含的遗传多样性揭示了编码自然产物中化学多样性的新水平。近年来,随着测序技术的不断进步,大量不同环境条件下的宏基因组数据也陆续被报道。2008年,研究者利用海洋环境中的宏基因组样本,获得了具有生物技术潜力的蛋白质编码基因,从而能够比以前更大程度地利用巨大的、尚未开发的海洋微生物多样性资源[4]。2009年,美国国立研究院发起的人类微生物组计划(HMP)公布了大量高质量宏基因组数据供科学界使用,宏基因组数据的产生使人们获得的信息量更多,并且取代了16S rRNA 数据成为新的研究热点,但是就当时而言,处理和分析此类数据的软件并不成熟[5]。由于宏基因组学研究的兴起,越来越多的研究者致力于开发用于数据分析的软件和工具,并在准确性和精确度上有了很大的提高。宏基因组数据的分析使人们对微生物的多样性、群落功能以及微生物与环境之间的关系有了更全面的了解。随着高通量测序技术和生物信息学的发展,微生物宏基因组数据分析的研究方法已经十分丰富,其基本流程如图1所示。本文着重讨论了宏基因组数据分析的方法及使用软件,具体内容主要包括以下六个部分:质量控制、组成分析、序列组装、序列分箱、基因预测和功能分析。系统概述了当前微生物宏基因组数据分析的基本流程和使用软件(表1),对宏基因数据分析工具及软件的优缺点和适用范围进行了总结,为研究者快速筛选分析方法,揭示数据背后的生物学意义提供参考。
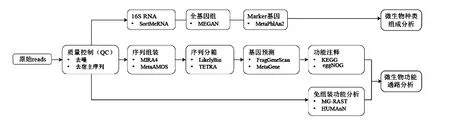
图1 宏基因组数据分析流程图Fig.1 Workflow for metagenomic data analysis

表1 宏基因组数据分析相关软件
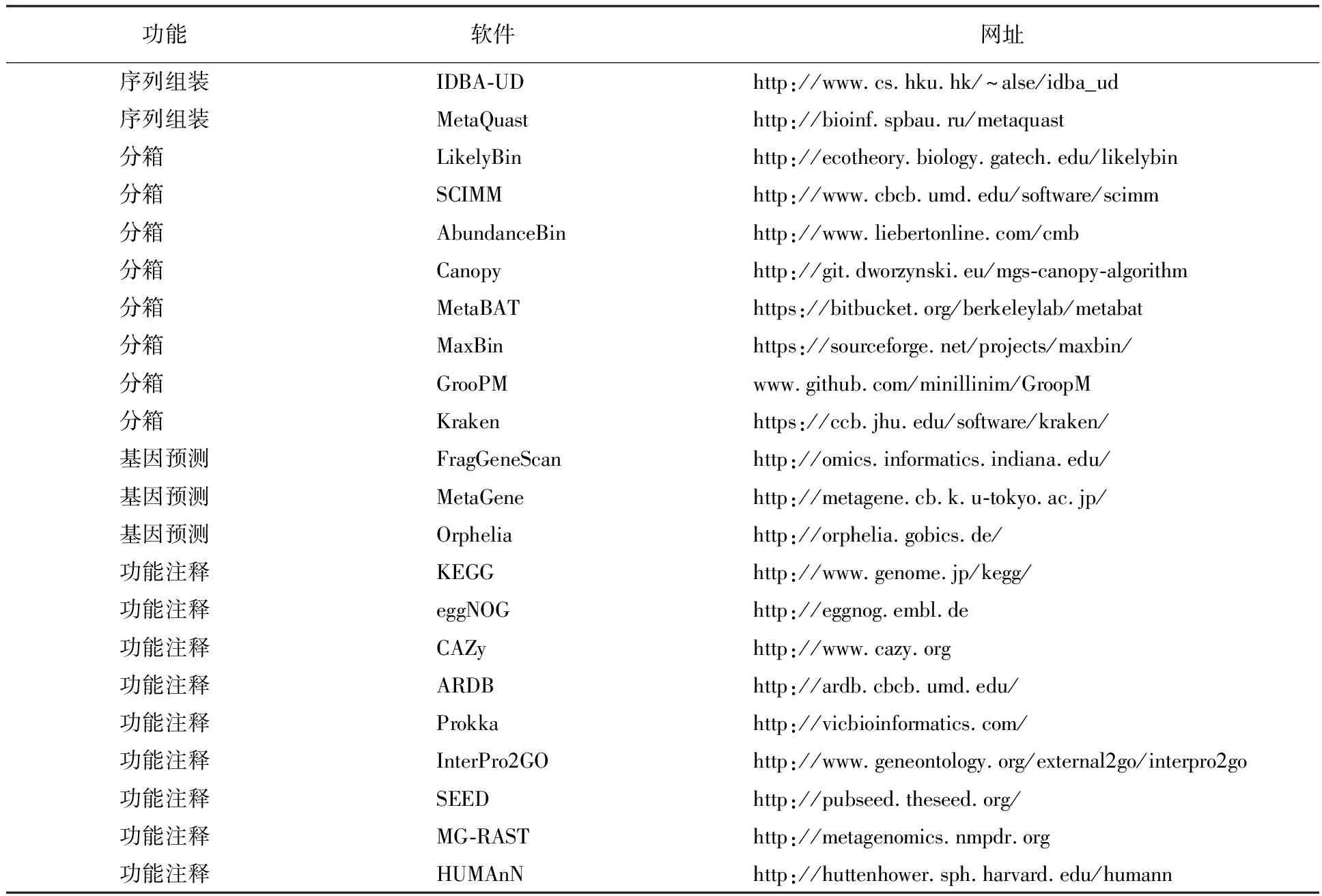
续表1
1 质量控制
宏基因组的测序数据是由大量来自不同微生物的短reads组成,进一步分析前需要对序列进行质量控制(quality control, QC)。质量控制主要包括两个部分:去噪和去宿主序列。去噪是指剔除低质量的碱基和残留的人工序列,这些序列会降低微生物组成分析的准确性,影响denovo组装[6]。目前常用于去噪的软件包括Trimmomatic[7]、PRINSEQ[8]、FASTX-Toolkit等。Trimmomatic是一种更加灵活、高效的预处理工具,它可以剪切引物、低质量的碱基和序列,并将序列剪切到一定长度且不干扰下游序列的分析,是一个专门针对Illumina的输出而设计的工具;PRINSEQ除了可以剪切序列和碱基外,还可以通过GC含量过滤序列,对序列进行汇总统计;FASTX-Toolkit与Trimmomatic的质量控制功能相似,FASTX-Toolkit可以对序列进行汇总统计,但是产生结果的质量不如Trimmomatic。
宿主序列会引起微生物序列组装错误,影响分析结果的准确性。Ward 等[9]在人的母乳和粪便样本中,发现人类DNA序列分别达到了宏基因组文库的64%和77%,这也进一步说明了去宿主序列的必要性。常用的工具包括MG-RAST[10]、TopHat2[11]等。MG-RAST 是基于Web界面分析的软件,而TopHat2是利用计算机将序列映射并与参考基因组比对的软件,需要有较高计算能力的计算机,而MG-RAST不需要。这两种软件均可用于牛瘤胃微生物宏基因组和转录组中DNA和RNA的过滤。
2 微生物组成分析
宏基因组数据分析主要包括组成分析和功能分析。微生物组成分析是基于宏基因组或转录组进行分类,并进一步分析微生物的丰度、种类等信息。对微生物种类分析主要有三种方法:16S RNA分析、全基因组分析、Marker基因分析。16S RNA分析可以有效地反应不同微生物群落的组成特征。SortMeRNA[12]是基于16S RNA分类的软件,可以快速整理所有与rRNA数据库匹配的片段并对多个序列进行排序,但是它很难检测低丰度的微生物,分析过程也较为复杂。全基因组包含了微生物菌群的所有遗传信息,通过全基因组分析不仅可以揭示微生物的组成特征,还能获得相关的功能信息,如MEGAN[13]是一个被广泛应用的软件,它为宏基因组、宏基因转录组、rRNA提供了一种综合的方法,可以执行多个数据库的比较,并通过使用InterPro2GO[14]、SEED[15]、KEGG等软件进行功能分析。Marker基因分析对原始物种水平进行了补充,包括真核、病毒的定量系统,菌株水平的鉴定以及菌株的追踪,但严重依赖于选择的Marker基因并且不能将物种与功能联系起来,也无法重构基因组进行组装,如MetaPhlAn2[16]使用“家族特异”的Marker基因对物种进行分类。Marker基因包括细菌、古菌、真核和病毒,可以实现精准的分类群分配、准确的估计物种的相对丰度和种水平精度,并以超快的分析速度实现株水平的鉴定和追踪。
3 宏基因组序列的组装
宏基因组序列的组装是进行功能分析前重要的步骤。通过测序技术得到的reads较短,需要组装获得更长的contigs[17],如果contigs序列过短且出现错误,会进一步影响序列分箱、基因预测和功能注释的准确性。序列组装一般包括以下步骤:首先输入一组短的基因序列,通过序列比对计算每对序列的重叠率,将较短的序列合并至较长的序列。目前主要有两种组装方式:依赖参考序列的组装和从头(denovo)组装。
3.1 依赖参考序列的组装
依赖参考序列的组装是用一个或者多个已知的基因组为参考进行组装的方法,但是如果序列存在插入、缺失或拷贝数的变化,便无法使用该方法组装。常用的软件包括MIRA4[18]、MetaAMOS[19]。MIRA4可以检测和分类单核苷酸变态性(SNPs),组装大量高度相似的序列,具有高度特异性和敏感性,但是该软件只用于mRNA转录样本。MetaAMOS可以检测基因并识别变异序列,减少组装宏基因组样本时的装配误差,比MIRA4准确性更高、成本更低。但当宏基因组数据来自新的环境时,受参考基因组数据库覆盖能力所限制,这些软件表现不佳[20]。
3.2 de novo组装
在没有已知参考基因组信息的情况下,将序列从头组装的过程即为denovo组装。该组装方式可以克服未知的瘤胃微生物基因组的瓶颈,因此普遍应用于瘤胃宏基因组的研究。目前新的组装软件不断被推出,组装的准确性和性能也有所提升。例如:MetaVelvet[21]、Meta-IBDA[22]。Meta-IBDA除可重建较长的contigs外,还可对来自同一物种不同亚种相似的contigs进行比对,这一特点可用来研究不同亚种基因组的结构变异和确定具有亚种生物学功能的保守区。从模拟的数据结果来看,MetaVelvet在目、科、属等水平上N50评分均高于Meta-IBDA,Meta-IBDA在种水平上特异性较高,整体来看MetaVelvet的表现优于Meta-IBDA。
4 宏基因组序列的分箱
宏基因组学分箱(binning),即将宏基因组测序片段按照物种、亚种或者属进行分类。分箱是根据基因特征对contigs或scaffolds进行分组,并对重建的基因组进行可视化。分箱的结果不仅可用于物种分类评估,还可以研究基因组内基因间的关联性,直接影响宏基因组学研究的深度和精确度[23]。根据用于对现有序列进行分组的信息,宏基因组分箱可以分为两种,即基于参考数据库分箱和无参考数据库分箱。
基于参考数据库分箱的原理是与参考数据库中的序列比对,对序列进行分类。Huson等[24]开发的 MEGAN是最早使用该原理的分箱软件。使用该软件分箱之前应利用BLAST[16]或其他比对工具将基因序列与已知序列数据库进行比较,然后使用MEGAN计算和探索数据集的分类内容,最后用NCBI对结果进行汇总和排序。但由于现有的数据库中物种数量非常少,参考数据库还不够完整、局限性较大、计算时间难以把握,此类方法对新环境中的序列样本具有很大的局限性[25]。
无参考数据分箱是应用聚类技术对序列进行分类,可直接处理样本[26]。根据现有的技术可以分为三类:基于序列组成的方法、基于丰度的方法和混合的方法[27]。基于序列组成的方法主要应用于具有不同核苷酸组成基因型的群落,并利用寡核苷酸频率和基因组碱基组成的特征进行分类[31]。该方法虽然避免了计算上常见的难题,但是在短reads执行上有一定的限制,常用的软件有LikelyBin[28]、TETRA[29]和SCIMM[30];基于丰度的分箱技术与基于序列的分箱技术相似,主要区别在于前者聚类的形成是由k-mer丰度来定义的,而不是由序列组成。该方法可以对短reads分箱,但是准确度不高,常用的软件有AbundanceBin[31]和Canopy[32];混合方法是将上述两种方法组合在一起的新技术,有望获得最佳的结果。Lin等[33]证明,结合反映物种丰度的序列组成信息和丰度信息,可以获得比其他两种方法更准确的分箱结果,相关软件有MetaBAT[34]、MaxBin[35]和GroopM[36]。
5 宏基因组基因预测和功能注释
宏基因组基因预测和功能注释是宏基因组数据分析的重要环节,通过基因预测和功能注释,可以获得相关的代谢通路信息[37]。基因预测是利用预测工具在基因组文库中识别潜在的开放阅读框(ORF)并鉴定蛋白质编码序列,预测结果的准确性受多个因素的影响,尤其是测序的误差和reads的长度。目前针对基因测序常用的软件有FragGeneScan[38]、 Orphelia[39]和MetaGene[40]。Trimble等[41]通过统计模拟数据对这些软件做了比较,FragGeneScan 对包含错读、低质量序列的识别具有较高的灵敏度,是目前可用的最准确的基因预测软件之一,而Orphelia和 MetaGene适合更高质量的序列。因此,用户可以根据测序数据的特点选择合适的方法。
功能注释是将基因或蛋白序列在特定的功能数据库中进行比对,将基因或蛋白与特定的功能联系起来,帮助了解相关的代谢通路,并进一步理解宿主与菌群、宿主与环境之间的关系。常用的功能注释数据库包括KEGG[42]、eggNOG[43]和CAZy[44]等。 KEGG包含三个类型的数据库:PATHWAY数据库、KO数据库和COMPOUND数据库,分别整合了蛋白质、基因组和化学功能信息,尤其在PATHWAY数据库中还包含了细胞代谢、遗传和细胞周期信息等,提供了一个更全面直接的分析方法;eggNOG提供了最全面和最新的数据库,它所提供的蛋白质功能注释的1 133个基因组跨越了三个领域,该数据库最重要的一个特征是OGs自动功能分析和功能描述的注释;CAZy是碳水化合物活性酶数据库,它将序列与分解寡糖、多糖的酶特异性和三维结构联系起来,提供了一个在线不断更新的家族分类方法。目前功能注释的软件越来越多,技术方面也逐渐成熟,研究者可根据研究的代谢途径选择相应的注释软件。
6 宏基因组免组装功能分析
免组装功能分析是将原始reads(QC后)与现有功能数据库或带注释的参考基因组进行比对获得功能信息的方法。该方法的准确性依赖于参考数据库的完善程度。对于已知的环境,如人的肠道,有关人肠道微生物的数据库已经十分完善,所以这种方法获得的结果是可信的。但是对于没有完整参考数据库的环境,如瘤胃(未分类和命名的微生物占比大于55%),这种方法获得的结果就有很大的局限性[45]。MG-RAST[10]和HUMAnN[46]是目前被普遍应用的软件。MG-RAST可供研究者免费使用,该软件不受特定基因组和数据类型的限制,目前已有500个宏基因组通过该软件处理。HUMAnN可直接从短序列中确定群落中的基因家族和存在的代谢通路及其相对丰度,能准确、有效地描述微生物代谢途径和生物学功能。人类微生物组计划(HMP)中就使用该软件研究了来自102个人中7个主要身体部位的649个宏基因组,并确定了24个普遍存在的代谢途径。
7 展 望
随着测序技术的发展,宏基因组测序的微生物组研究已渗透到各个领域,包括海洋、土壤、人、动物以及环境等。通过对宏基因组数据的分析,挖掘了微生物的多样性、种与种之间的进化关系,揭示了微生物与环境、微生物与宿主之间的协作关系。但是,宏基因组数据分析最大的难题仍是生物信息学和计算瓶颈,如序列组装。由于宏基因组数据比较复杂,在组装过程中高度相似的序列容易出现错误,组装和分箱的准确率不高,而且计算成本较高,因此软件性能等各个方面还有很大的提升空间。一方面要加强实验和分析流程的标准化,另一方面提高对未知环境的微生物组研究和认识,将已有的数据整合构建为高质量的参考数据库,其次,优化宏基因组数据的组装和分箱流程,开发新的算法,提高宏基因组数据的利用率。
猜你喜欢
中国人兽共患病学报(2022年9期)2022-10-19
军事文摘(2022年16期)2022-08-24
今日农业(2021年11期)2021-08-13
科学导报(2021年29期)2021-06-03
中国生殖健康(2020年4期)2021-01-18
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
科海故事博览·下旬刊(2019年6期)2019-04-16
财经(2017年2期)2017-03-10
财经(2016年15期)2016-06-03
