
TRPC6基因新发变异致儿童终末期肾病1例报告并文献复习
2020-12-23 06:54樊姣姣和俊杰刘雪杰王淑静同少峰
临床儿科杂志 2020年12期
樊姣姣 付 荣 和俊杰 刘雪杰 陈 冲 王淑静 同少峰
新乡医学院附属濮阳市油田总医院儿科(河南濮阳 457001)
终末期肾病(end-stage renal disease,ESRD)是各种因素所致慢性肾脏病的最严重阶段,肾小球滤过率(glomerular filtration rate,GFR)<15 mL/(min·1.73 m2),需要透析或肾移植等替代治疗维持生命[1]。遗传学研究表明,编码足细胞相关蛋白基因发生变异可破坏肾小球正常的滤过膜结构,导致蛋白尿的发生和进行性肾功能衰竭,如NPHS1,NPHS2,PLCE1,ADCK4,WT1等[2]。现回顾性分析濮阳市油田总医院诊治的1例由于TRPC 6基因新发变异导致临床表现严重且短时间内进展至ESRD患儿的临床资料,并对相关文献进行复习。
1 临床资料
先证者,女,9岁,因发现颜面水肿20天就诊于当地医院,尿常规蛋白+++,24小时尿蛋白总量2 677 mg/L;血白蛋白15.7 g/L,三酰甘油4.59 mmol/L,总胆固醇10.66 mmol/L,尿素氮5.94 mmol/L,肌酐68 μmol/L,GFR 86 mL/(min·1.73 m2),乙肝病毒相关抗体及抗核抗体谱均阴性。患儿父母体健,非近亲婚配;有一姐姐,体健;否认家族性肾脏疾病及其他遗传性疾病史。外院诊断为原发性肾病综合征,曾采用中药制剂、激素及呋塞米利尿消肿及白蛋白提高胶体渗透压等治疗,患儿水肿无好转,遂至上级医院就诊。入院体格检查:颜面部浮肿,腹部叩诊移动性浊音阳性,双下肢凹陷性水肿,外阴水肿。实验室检查:24小时尿蛋白总量4201 mg/L;血白蛋白16.1 g/L,总胆固醇20.52 mmol/L,三酰甘油4.44 mmol/L,尿素氮 19.39 mmol/L,肌酐95.8 μmol/L,GFR 62 mL/(min 1.73 m2);腹部B超示左肾102 mm×54 mm×53 mm,右肾108 mm×43 mm×42 mm,双肾实质弥漫性回声改变,腹腔大量腹水。予中药制剂,激素冲击治疗,口服免疫抑制剂(他克莫司/环孢素A),输注白蛋白、血浆等治疗,但是患儿治疗反应差,全身高度水肿,尿量减少至300~400 mL/d,体质量自入院22 kg增加至32 kg。2周后复查尿常规蛋白+++;血尿素氮47.97 μmol/L,肌酐247.0 μmol/L,GFR 23 mL/ (min·1.73 m2),提示肾功能持续进行性恶化。行血液透析治疗后,患儿水肿逐渐消退,出院后规律血液透 析。
5个月后患儿因呼吸急促3小时第1次入油田总医院儿科。入院体格检查:精神差,贫血貌,呼吸急促,三凹征阳性,全身皮肤黏膜暗黄,双眼睑水肿,双肺呼吸音粗,左肺可闻及细湿啰音,肝肋下2 cm,质软,移动性浊音阴性,双下肢凹陷性水肿。实验室检查:尿常规蛋白+++;外周血白细胞22.63×109/L,血红蛋白78 g/L,白蛋白25.6 g/L,血钾6.53 mmol/L,尿素氮18.55 mmol/L,血肌酐797.8 μmol/L,GFR 7.5 mL/(min·1.73 m2),B型尿钠肽>5 000.0 ng/L;心脏彩超示心包积液,右侧胸腔积液,肺实变。诊断:慢性肾脏病5期、心力衰竭、肺部感染、肾性贫血、高钾血症,予抗感染、输血、床旁血液透析等对症支持治疗。治疗18天后患儿病情好转,回当地医院继续规律透析。
因频繁抽搐,呈全身强直阵挛发作,再次入总医院儿科。入院体格检查:精神差,嗜睡,肝肋下3 cm,质软,双侧巴氏征弱阳性。实验室检查:血白蛋白31.5 g/L,尿素氮16.32 mmol/L,肌酐628.5 μmol/L,GFR 9.7 mL/(min· 1.73 m2),血钙2.23 mmol/L,血钾3.80 mmol/L;头颅核磁共振(MRI)示大脑半球对称,脑实质未见明显异常信号,脑室系统扩大,中线结构居中,脑沟、脑池稍增宽,余未见异常。视频脑电图:广泛性棘慢波、多棘慢波、棘波节律发放。诊断:慢性肾脏病5期、癫痫,予血液透析,口服丙戊酸钠和左乙拉西坦等。经治疗后,患儿病情趋于平稳,自动出院后电话随访,未再出现痫样发作。
考虑患儿临床治疗效果差,进展至ESRD速度快,出现心力衰竭和癫痫原因不明,经医院伦理委员会批准,患儿父母签署知情同意书后,采集患儿外周血2 mL提取基因组DNA进行全外显子基因测序(whole exon gene sequencing,WES),然后取患儿及父母外周血进行Sanger测序,验证WES筛选出的致病基因是否存在常染色体隐性或显性遗传的可能。测序实验委托北京智因东方转化医学研究中心有限公司完成。基因检测结果显示,患儿TRPC6基因第2个外显子发现c.395T>C(编码区第395位碱基胸腺嘧啶转换为胞嘧啶),导致蛋白质第132位蛋氨酸转换成苏氨酸(p.Met132Thr),为错义变异(图1)。该氨基酸在物种进化过程中高度保守,变异后可能影响蛋白质的功能和表达,先证者父母经Sanger测序验证均未见异常,为新发(de novo)变异。根据美国医学遗传与基因组学会(American College of Medical Genetics and Genomics,ACMG)指南进行生物学致病等级分析,保守性及蛋白结构(SIFT、Polyphen2_HDIV、Mutation_Taster)两种统计方法预测变异有害,可能致病;该变异的次要等位基因频率(MAF)<0.005,属于低频变异,dpSNP、千人基因组、ExAC等数据库未见收录,且该变异位点的致病性已有文献报道,但为国内首次发现[9,13]。
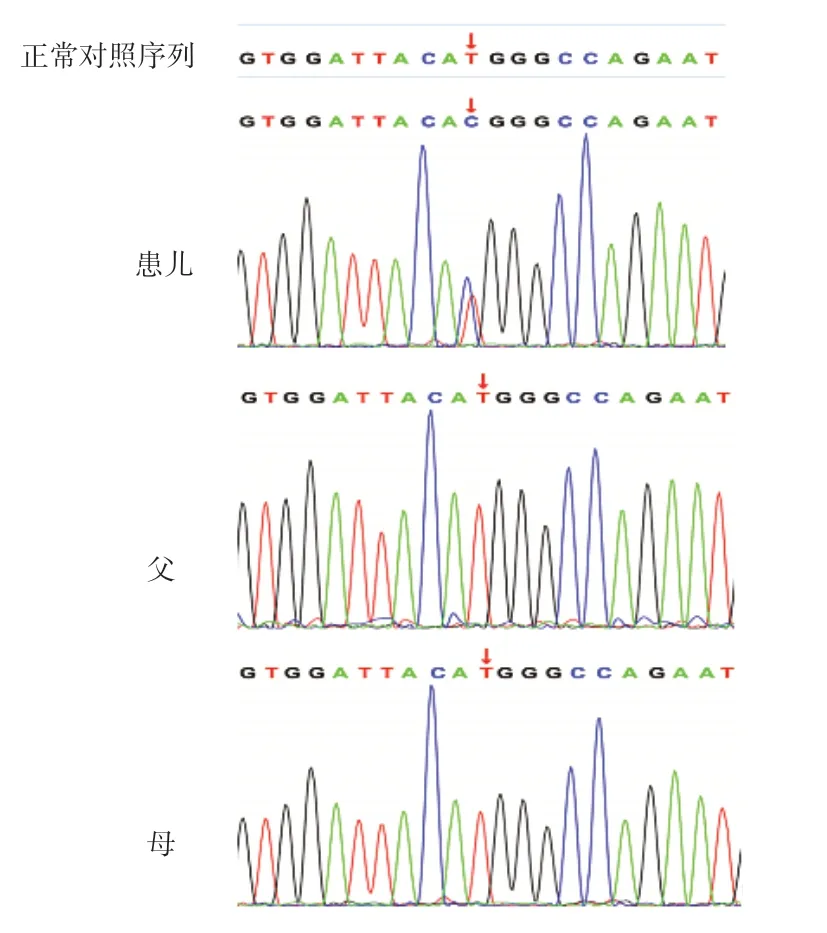
图1 患儿及父母TRPC6 基因测序图
2 讨论
TRPC 6基因编码瞬时受体电位阳离子通道 6蛋白(transient receptor potential channel 6,TRPC6),是一个非选择性阳离子通道,位于染色体 11q21~q22,包含 13个外显子,共 132 365个碱基。TRPC6表达增加会导致足细胞钙离子分布异常,改变足突的收缩功能,破坏滤过屏障的动态平衡,导致蛋白尿的发生;也可导致足细胞肌动蛋白骨架结构异常,引起肾小球的硬化[3-5]。2005年研究者在常染色体显性遗传的家族性局灶节段性肾小球硬化(focal segmental glomerulosclerosis,FSGS)中检出并证实TRPC6为致病基因,且60%患者在约10年间进展至ESRD[6]。同年,研究者通过大鼠肾脏切片发现TRPC 6在肾小管和肾小球广泛表达,多数分布于足细胞,尤其是裂孔隔膜,并对71个FSGS家系进行TRPC6筛查,变异检出率约为7%,发病年龄较晚(17~57岁)[7]。2009年首次在1例9岁FSGS患儿中检出TRPC6基因(c.2339T>C)变异,并认为TRPC6基因也可导致散发性的早发型FSGS。
迄今为止,已报道儿童TRPC6基因的致病位点变异16例,包括错义变异14例、无义变异1例和移码变异1例,无明显的热点变异位点。但在对伊朗26例<16岁的FSGS患儿的TRPC6基因筛查中,新发现4种变异位点,其中3例错义变异位于外显子2,并认为第2外显子应该作为热点筛查区域[8]。回顾文献报道的18例患儿[8-15],加上本例患儿共19例,发病年龄最小4月龄,最大14岁,平均年龄(5.48±3.92)岁。18例报道患儿中,3例未报道病理类型,12例是FSGS。本例患儿由于大量腹水和全身高度水肿,未能行肾穿刺活检术,因而其病理类型未知;18例报道患儿中5例出现肾衰竭,进展至ESRD时间最短为1年,最长13.8年,且均无肾外表现[8-15]。
本例患儿的临床表现严重,在病程2月余出现少尿,全身高度水肿,尿素氮和血肌酐迅速增高,应用Schwartz公式估算的GFR从86 mL/(min·1.73 m2)下降至23 mL/(min·1.73 m2),说明肾功能急剧恶化[16],并且对激素、利尿剂、免疫抑制剂等治疗反应差,需进行血液透析。国外已有2篇文献报道TRPC6基因错义变异c.395T>C(p.Met132Thr)导致儿童FSGS,与本例患儿临床表现的相同之处是均进展至ESRD[9,13],推测该变异位点导致儿童肾衰竭的频率较高,需引起临床医生的高度重视;不同之处是进展至ESRD的时间分别是9.5年和1年,且未见肾外表现受累的描述,然而本例患儿进展至ESRD的时间约为6个月,并出现循环和神经等肾外系统受累的表现,表明TRPC6基因同一变异位点的临床表现异质性较大,提示临床治疗需个体化并注意监测肾功能[10,13]。
本例患儿在病程7月余出现心力衰竭,分析原因认为除了与贫血(血红蛋白78 g/L)、感染(外周血白细胞22.63×109/L)、高钾血症(血钾6.53 mmol/L)、水钠潴留、透析不充分等与多种导致ESRD可并发心力衰竭的危险因素相关以外,还可能与TRPC6基因变异相关[17-19]。研究发现,TRPC6在小鼠心肌中特异性过表达导致胞内钙超载,可诱发心肌肥大和心力衰竭[20];另有研究证实,TRPC6在心室肌细胞的蛋白表达增加,可促进心肌细胞的凋亡[21]。研究记录细胞电流显示,M132T变异体的内向Ca2+电流幅度是野生型的10倍,并且与其他突变体相比,M132T变异体的离子通道在100 s内没有任何失活现象,可能导致严重的钙内流[10]。本例患儿同样携带M132T变异,故推测细胞内Ca2+浓度的大幅增加是导致心肌细胞损伤和心力衰竭的发生的重要因素。
本例患儿在病程8月余出现继发性癫痫,呈全身强直-阵挛发作,持续时间长,次数频繁,考虑原因也与TRPC6基因变异有关。TRPC6基因在脑组织对神经元的正常发育、树突的生长和树突棘的形成等神经网络的构建十分重要,该通道的稳定表达可调节神经元的兴奋性,而细胞内过度Ca2+超载,会导致神经元的异常放电和功能紊乱,提示TRPC6蛋白的异常表达可能与癫痫的发生密切相关[22]。TRPC6基因致痫的分子机制尚不明确,但研究表明,在难治性颞叶癫痫患者的脑皮质和毛果芸香碱诱导的癫痫状态的模型小鼠海马中发现,TRPC6蛋白的表达明显升高,然而在脑室注射TRPC 6抗体可降低树突分支的数量和树突棘的密度,从而引发突触网络的重建,TRPC6通道可能是癫痫相关突触重构的致病因素[23];在导致顽固性癫痫病因之一的局灶性皮质发育不良患者皮质组织中发现TRPC6的蛋白表达明显升高,促进树突细胞的生长和树突棘及兴奋性突触的发育,提示与癫痫的发生有关[24];在结节性硬化症癫痫患者病灶皮层脑组织中TRPC6表达上调,提示异构神经元的特异性分布模式参与了致痫机制[25]。本例患儿携带M132T变异可导致细胞内Ca2+浓度过度升高,且通过口服丙戊酸钠和左乙拉西坦两种钙通道阻滞剂的抗癫痫药物后痫性发作得到有效控制。故推测,TRPC6基因变异可能是本例患儿出现癫痫发作的主要原因。
近年来研究者先后在中国激素耐药型肾病综合征患儿检测到TRPC6基因变异,但患儿在随访过程中肾功能均未进展至ESRD[11,15]。通过WES测序技术明确本例临床表现严重、肾功能快速恶化、进展至ESRD时间短、出现循环和神经等肾外系统受累患儿的致病原因是TRPC 6基因新发变异c.395 T>C(p.Met132Thr),国内未见报道,丰富了TRPC6基因型与临床表型。
猜你喜欢
心电与循环(2021年4期)2021-11-29
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
中学数学杂志(2019年9期)2019-05-29
饮食科学(2017年5期)2017-05-20
中国眼镜科技杂志(2016年17期)2016-10-24
安徽医科大学学报(2015年9期)2015-12-16
百科知识(2015年18期)2015-09-10
西南军医(2015年4期)2015-01-23
中国中医药现代远程教育(2014年20期)2014-03-01
