
建神曲发酵过程中微生物群落结构及功能预测分析
2021-02-26 11:13廖茜
福建农业科技 2021年12期
关键词:高通量测序




摘 要:为明确建神曲发酵过程中的微生物种类、优势群落组成及功能特征,采用高通量测序和分析技术对不同发酵时间点的建神曲细菌菌群结构和基因功能进行分析。结果表明,建神曲发酵过程中的细菌种类较少,主要由Firmicutes、Proteobacteria、Bacteroidetes、Actinobacteria等4个门细菌组成,不同发酵阶段的建神曲共有1个核心菌群;在整个发酵过程中,优势菌群的丰度出现明显的变化趋势,表明建神曲的发酵过程存在清晰的细菌演替过程;PICRUSt基因功能预测分析结果显示,在丰度排前50的KEGG功能基因KOs中,有22个KOs与酶相关,有49个KOs的丰度在不同发酵时间点之间存在显著性差异(P<0.05),说明建神曲中的细菌含有丰富的与代谢途径相关基因,且不同发酵时间点的生理功能不同。
关键词:建神曲;发酵过程;高通量测序;细菌菌群结构;基因功能预测
中图分类号:Q 938 文献标志码:A 文章编号:0253-2301(2021)12-0007-05
DOI: 10.13651/j.cnki.fjnykj.2021.12.002
Analysis on Microbial Community Structure and Function Prediction Duringthe Fermentation of Jianshenqu
LIAO Qian
(Fujian Pharmaceutical Examination and Inspection Center, Fuzhou, Fujian 350001, China)
Abstract: In order to clarify the microbial species, dominant community composition and functional characteristics during the fermentation of Jianshenqu, the high-throughput sequencing and analysis techniques were used to analyze the bacterial community structure and gene function of Jianshenqu bacteria at different fermentation time points. The results showed that there were few bacterial species in the fermentation process of Jianshenqu, which were mainly composed of Firmicutes, Proteobacteria, Bacteroidetes and Actinobacteria. There was one core flora in Jianshenqu at different fermentation stages. During the whole fermentation process, the abundance of the dominant flora showed an obvious trend of change, indicating that there was a clear bacterial succession process in the fermentation process of Jianshenqu. The results of PICRUSt gene function prediction analysis showed that among the KEGG functional gene KOs with the top 50 abundances, 22 KOs were related to enzymes, and the abundance of 49 KOs was significantly different at different fermentation time points (P<0.05), indicating that the bacteria in Jianshenqu contained abundant genes related to metabolic pathway, and the physiological functions were different at different fermentation time points.
Key words: Jianshenqu; Fermentation process; High-throughput sequencing; Bacterial community structure; Gene function prediction
建神曲,產自福建泉州,又名泉州神曲、范志曲、百草曲,主要为面粉、小麦、麸皮与砂仁、广陈皮、广藿香等药物混合后发酵而成的加工品[1]。建神曲肇始于明、清年间,迄今已有400多年历史,在《本草纲目拾遗》《蔡氏药贴》《药性考》等古今中医书中均有详载[2]。建神曲主要用于伤食胸痞,腹痛吐泻,痢疾、外感风寒,小儿伤饥失饱以及旅行中水土不服而引起的肠胃不和。《本草纲目拾遗》曰:气味中和,清香甘淡,能搜风解表,开胸块隔,调胃健脾,消积进食,和中解酒,止泻利水,治四时不正之气,感冒发热,头眩咳嗽及伤食腹痛,痞满气痛,呕吐泻泄痢疾,饮食不进等症[3]。
根据《福建省中药饮片炮制规范(2012年修订)》记载,建神曲的制法是将麸皮、面粉、小麦与砂仁、广陈皮、广藿香等药材经处理后混匀,捣软压模制成长方形小块,蒸1 h,经自然发酵后,取出晒干或低温干燥,刷净外表,包装即得。从现代微生物学的角度来看,建神曲发酵过程的实质为微生物生长、演替、互作和代谢的过程,通过微生物和酶的催化分解作用,使药物发泡、生衣,产生微生物次生代谢产物、消化酶、低聚糖等新的活性成分协助消化系统发挥正常功能[4-5]。自然发酵工艺由于不同发酵周期的温湿度条件存在差异且发酵时间较长,造成参与发酵的微生物的数量和种类不确定,会导致同一产地不同批次的产品质量存在差异。利用优势菌种建立现代混合发酵炮制工艺,可以改善自然发酵过程杂菌多、时间长、产品质量不稳定的弊端[6]。但目前对建神曲发酵过程中的菌群结构组成的研究报道较少。因此,本研究采用高通量测序和分析技术明确建神曲发酵过程中的微生物群落结构和功能特征,为后续建神曲建立稳定可控的现代发酵工艺提供参考依据。
1 材料与方法
1.1 样品采集
本试验建神曲样品采自泉州中侨药业有限公司的“老范志万应神曲”,发酵周期约为12 d。本试验选择发酵前(0 d)、发酵中期(6 d)和发酵结束(12 d)时进行取样,编号分别为D0、D6、D12。每个取样时间点分别随机取3个建神曲长方形小块,为减少外界环境杂菌的干扰,用无菌手术刀切取建神曲长方形小块中心样品2~3 g,切下的样品放置于-80℃下保存备用。
1.2 DNA提取与PCR扩增
保存的建神曲样品DNA采用土壤微生物DNA提取试剂盒(Mobio,USA)提取,提取步骤依据试剂盒操作说明书进行。提取后的基因组DNA采用16S rRNA基因V3~V4区片段通用引物对341F和805R扩增。扩增后的PCR产物用PCR纯化试剂盒(Qiagen, Germany)纯化,再用Qubit 3.0 荧光定量仪(Invitrogen, USA)进行定量。
1.3 IlluminaMiSeq测序与高通量数据分析
纯化后的PCR扩增产物进行Illumina MiSeq平台高通量测序,由生工生物工程(上海)股份有限公司完成。测序得到的高通量原始数据先去除测序时加入的正反向引物[7],再根据序列之间的overlap关系将成对的reads拼接成一条完整的序列[8],对序列进行质控过滤[9]。得到的有效序列再去除嵌合体和冗余序列以及划分分类操作单元(OTUs)[10],将OTU代表序列与Ribosomal Database Project(RDP)数据库进行比对进行物种注释。利用Mothur软件和QIIME软件对生物样本的alpha多样性和beta多样性进行分析[11-12]。
1.4 数据统计分析
在本研究中平均值、标准偏差和P-values采用SPSS软件计算,多重比较分析采用Tukey′s HSD检验;柱状图、韦恩图和热图采用R软件绘制。
2 结果与分析
2.1 高通量測序数据分析
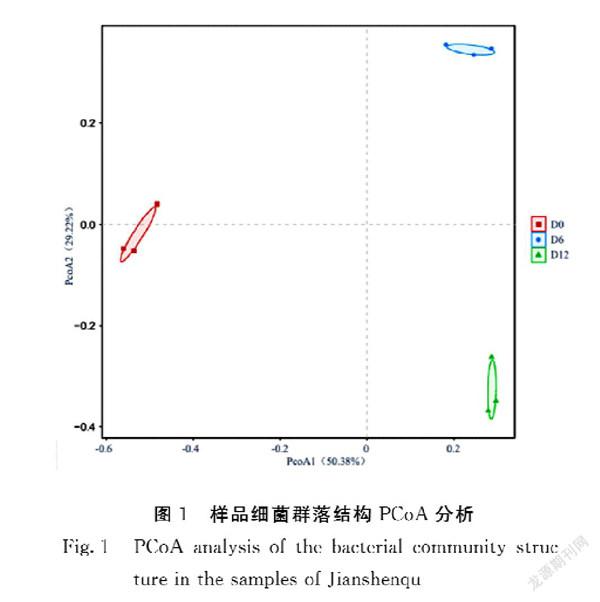
从表1可知,3个发酵时间点的9个建神曲样品获得的16S rRNA基因有效序列数为48372~67995条,OTUs数为156~199个且OUTs覆盖率(Good′s coverage)均>99%,表明建神曲中的细菌种类较少,且高通量测序获取的数据量能够真实地反映样品的细菌多样性情况。Alpha多样性统计分析结果表明,发酵前和发酵结束样品中的Simpson指数显著高于发酵中期样品(P<0.05),而发酵前和发酵结束样品中的Shannon指数显著低于发酵中期样品(P<0.05),表明建神曲在发酵前和发酵结束时的样品细菌个体数目在群落中分配的均匀程度比发酵中期差,也反应建神曲发酵过程存在细菌优势菌群的演替过程。根据Beta多样性距离矩阵进行PCoA分析,结果(图1)表明相同发酵时间点样品的微生物结构和组成十分相似,不同发酵时间点样品的微生物组成结构存在明显差异。
2.2 建神曲发酵过程中细菌群落结构特征及演替分析
在门水平,建神曲主要由Firmicutes、Proteobacteria、Bacteroidetes、Actinobacteria等4个细菌门组成(图2)。从整个发酵过程来看,4个细菌门的丰度都出现明显的变化,发酵前期与发酵结束时4个细菌门的差异均达到显著(P<0.05)或极显著水平(P<0.01)。发酵前期最优势菌门Proteobacteria的丰度逐渐下降,次优势菌门Firmicutes的丰度逐渐上升,到发酵结束时建神曲中的最优势菌门变为Firmicutes。
在属水平,建神曲中主要由Lactobacillus、Pseudomonas、Bacillus、Pseudoxanthomonas、Pediococcus、Streptomyces、Flavobacterium、Weissella、Acinetobacter、Clostridium_sensu_stricto、Sphingobacterium、Paenibacillus、Chitinophaga、Comamonas、unclassified_Alcaligenaceae等12个细菌属组成(图3)。发酵前期最优势菌群为Pseudomonas(75.43±12.34)%,发酵中期和发酵结束时下降为(2.32±1.31)%和(6.12±1.32)%;发酵中期的优势菌群为Lactobacillus、Pseudoxanthomonas和Pediococcus,其丰度分别达到(25.62±14.58)%、(25.55±16.64)%和(19.96±9.02)%,发酵结束时Lactobacillus成为绝对优势菌属,丰度达到(71.98±12.16)%,说明整个发酵过程存在明显的细菌演替过程。
2.3 不同发酵时间点建神曲细菌群落组成比较分析
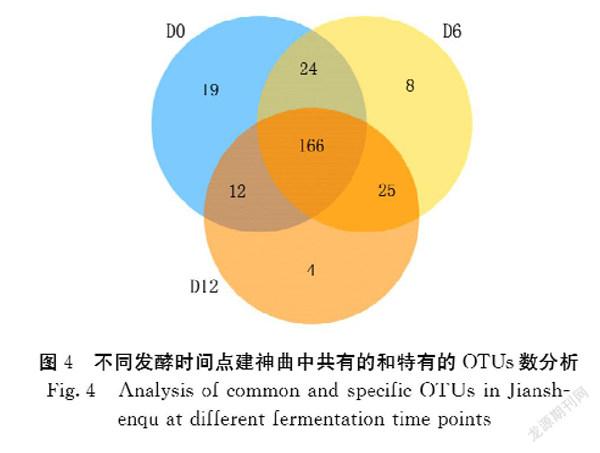
通过构建韦恩图对不同发酵时间点建神曲所共有的和特有的OTU数目进行分析(图4)。从分析结果可以看出,9个建神曲样品中细菌OTUs总共258个,其中共有OTUs数为166个,分别占发酵前期、发酵中期和发酵结束时OTUs总数的75.11%、74.44%和80.19%;而发酵前期、发酵中期和发酵结束时的建神曲中的特有OTUs数分别为19、8和4个,分别占OTUs总数8.60%、3.59%和1.93%;说明尽管不同发酵时期建神曲中的优势菌群不同,但不同发酵时期建神曲中的菌群种类基本一致。
2.4 建神曲细菌群落功能PICRUSt预测分析
对细菌的基因功能进行预测有助于理解建神曲中化学成分的转化过程。通过PICRUSt分析,9个建神曲样品共鉴定出5605个KEGG功能基因KOs,对应于41个2级功能层和328个3级功能层。选择丰度前50的KEGG功能基因KOs绘制热图(图5)。统计结果显示,50个优势KOs中有22个KOs与酶相关,且除K09687外剩余49个KOs丰度在不同发酵点均存在显著性差异(P<0.05);表明建神曲中含有丰富的与代谢途径相关的细菌功能基因以及不同发酵时间点微生物所起的功能不同。
3 结论与讨论
自然界中蕴藏着大量未被人们熟知的微生物资源,使用传统鉴定方法只能对可培养微生物进行鉴定,由于严格的专性厌氧环境和特殊的营养需求,使得其很难得到样本中的整个微生物结构组成情况[13]。随着高通量测序技术的发展,这一问题得到了解决。近几年,国内外学者通过高通量技术在多种环境样品中的微生物多样性方面都取得了初步进展[14]。本研究利用高通量测序技术对建神曲发酵样品中微生物菌落结构进行研究,确定了建神曲样品中的细菌主要由Firmicutes、Proteobacteria、Bacteroidetes、Actinobacteria等4个细菌门组成。从高通量测序数据分析,建神曲中的细菌种类相对简单,但核心菌群基本一致,这可能与建神曲中的药物成分对细菌物种存在自然选择作用有关。
优势菌种发酵技术是利用传统发酵菌种中的某种单一特定的有益菌株进行发酵,发酵时间短、容易形成规范化标准化发酵,同时可减少杂菌的产生[6]。从菌群结构和PICRUSt基因功能预测分析结果来看,各发酵时间点的建神曲细菌优势菌群和生理功能存在显著的差异(P<0.05),说明在建神曲发酵过程中不同阶段的细菌所起的作用不同;从差异变化趋势分析,发酵结束时的优势菌群在整个发酵过程中的丰度是逐渐增加的,说明建神曲在整个发酵过程中存在清晰的细菌演替过程,而优势菌群是需要在合适的环境下逐步演替显现出来。因此,如果仅采用某种单一的特定菌种进行发酵并不能达到混合菌种发酵的互补优势[5]。因此,分阶段探索建神曲中发酵过程中的优势菌株及功能,采用多种益生菌混合接力发酵的生产工艺,或能达到更好的效果。
参考文献:
[1]王炯,王子文.六神曲与建神曲之异同[J].西部中医药,2005,18(2):30.
[2]张子华.泉州传统名药——建神曲[J].海峡药学,1997,9(1):2.
[3]赵学敏.本草纲目拾遗[M].上海:人民卫生出版社,1983:88.
[4]陈彦琳,王云庭,关凯乐,等.六神曲发酵过程中微生物群落结构研究[J].中国中药杂志,2020,45(21):5219-5225.
[5]庞思奇,马嘉擎,林家慧,等.中药“六神曲”发酵工艺研究进展[J].食品与发酵科技,2021,57(4):113-116.
[6]贾丹丹.六神曲和百药煎炮制过程中微生物分离、鉴定与特性分析[D].上海:中国医药工业研究总院,2016.
[7]MARTIN M.Cutadapt removes adapter sequences from high-throughput sequencing reads[J].EMBnet Journal,2011,17(1):10-12.
[8]ZHANG J,KOBERT K,FLOURI T,et al.PEAR:a fast and accurate Illumina Paired-End reAdmergeR[J].Bioinformatics,2014,30(5):614-620.
[9]SCHMIEDER R,EDWARDS R.Quality control and preprocessing of metagenomic datasets[J].Bioinformatics,2011,27(6):863-864.
[10]EDGAR RC.UPARSE:Highly accurate OTU sequences from microbial amplicon reads[J].NatureMethods,2013,10(10):996-998.
[11]SCHLOSS P D,WESTCOTT S L,RYBIN T,et al.Introducing mothur:open-source,platform-independent,community-supported software for describing and comparing microbial communities[J].Applied and Environmental Microbiology,2009,75(23):7537-7541.
[12]CAPORASO J G,KUCZYNSKI J,STOMBAUGH J,et al.QIIME allows analysis of high-throughput community sequencing data[J].NatMethods,2010,7(5):335-336.
[13]曹荣,张井,孟辉辉,等.高通量测序与传统纯培养方法在牡蛎微生物群落分析中的应用对比[J].食品科学,2016,37(24):137-141.
[14]朱永官,沈仁芳,贺纪正,等.中国土壤微生物组:进展与展望[J].中国科学院院刊,2017,32(6):554-556.
(责任编辑:柯文辉)
收稿日期:2021-10-12
作者簡介:廖茜,女,1985年生,主管药师,主要从事药物微生物方面的研究。
猜你喜欢
江苏农业科学(2017年20期)2017-11-30
江苏农业科学(2017年16期)2017-10-27
湖北农业科学(2017年13期)2017-08-08
中国中药杂志(2017年13期)2017-07-31
山东工业技术(2017年12期)2017-07-06
中国中药杂志(2017年7期)2017-05-26
中国医药导报(2017年9期)2017-05-11
中国中药杂志(2016年24期)2017-04-18
中国中药杂志(2017年4期)2017-03-28
中国中药杂志(2017年3期)2017-03-20
