
糖环修饰的腺苷类似物的合成及其抗肿瘤活性研究
2021-02-28 03:40朱慧敏曾万波天津大学化工学院天津300350军事医学研究院毒物药物研究所抗毒药物与毒理学国家重点实验室北京100850北京解放军总医院第一医学中心心脏大血管外科北京100853
西北药学杂志 2021年6期
朱慧敏,曾万波,张 伟,李 东,徐 亮*(1.天津大学化工学院,天津 300350;.军事医学研究院 毒物药物研究所 抗毒药物与毒理学国家重点实验室,北京 100850;3.北京解放军总医院第一医学中心心脏大血管外科,北京 100853)
恶性肿瘤严重威胁人类健康,抗肿瘤药物的研发一直是研究热点。腺苷是人体内天然存在的内源性核苷分子,具有良好的安全性,临床已用于心律失常、心绞痛、心肌梗死等疾病的治疗。近年研究发现,腺苷及其类似物还具有潜在的抗肿瘤活性[1],可能与激活AMP依赖的蛋白激酶A(Adenosine 5′-monophosphate (AMP)-activated protein kinase,AMPK)[2]有关。
AMPK是生物体能量代谢调节的关键分子,肿瘤细胞的能量代谢途径已成为治疗肿瘤的潜在靶点[2]。正常细胞主要依靠线粒体的氧化磷酸化为细胞供能,而大多数肿瘤细胞则依赖有氧糖酵解,这种现象被称之为“Warburg” 现象,Brandon F等[1,3]研究发现,AMPK激活能够促进分解代谢途径,抑制合成代谢途径,并通过缺氧诱导因子(HIF-1)和肿瘤抑制基因p53降低肿瘤细胞糖酵解水平,抑制Warburg现象,从而抑制肿瘤细胞的生长。王钦加[4]研究发现,AMPK激活可以抑制哺乳动物雷帕霉素靶蛋白(mTOR)途径的异常活化,同时可通过乙酰辅酶A阳离子化酶(ACC)磷酸化促进能量代谢并诱导肿瘤细胞凋亡。
腺苷类化合物普遍半衰期短,不能有效穿过血脑屏障,胃肠道吸收差[5-7]。基于腺苷母核结构的修饰与改造是寻找效果更好、毒性更低、底物选择性更好的新型抗肿瘤药物的重要手段[8],例如8-氯环磷酸腺苷及其代谢产物8-氯腺苷均具有良好的抗肿瘤活性,目前都已进入临床试验阶段[9-10]。通过对8-氯腺苷亲脂衍生化和脂质体制剂手段的双重结合,可改善药物代谢动力学特性,并增加抗肿瘤活性和生物利用度[11]。通过取代的吡啶杂环替换糖基所得到的3-去氮腺苷类似物,具有良好的抗肿瘤细胞增殖活性[8]。2氮杂-ε-腺苷(la)增强了腺苷分子的亲脂性,在小鼠神经胶质瘤组织上表现出有效的抗肿瘤活性[12]。本研究以腺苷为先导结构,以增加亲脂性和结构多样化为目的,选择糖环羟基为修饰位点,通过2′ 和3′ 羟基取代成环以及共酯化的修饰策略,设计了一系列具有较好亲脂性的化合物,测试了这些化合物对MCF-7肿瘤细胞的抑制活性,并通过AutoDock Vina计算机分子对接模拟,探讨了优选化合物与AMPK的作用机制。
1 仪器与材料
1.1仪器 DF-101S型恒温加热磁力搅拌器和SHZ-D(Ⅲ)型循环水式真空泵,均购自巩义市予华有限责任公司;RE-52A型旋转蒸发器(上海亚荣生化仪器厂);GL-9406型手提紫外分析仪(海门市其林贝尔仪器制造公司);JNM-ECS400型核磁共振谱仪(日本电子株式会社);Agilent Technologies LC/MS 1200 series-6130 Quadrupole液质联用仪(美国Agilent Technologies 公司);Milli-O超纯水系统[美国密理博(Millipore)公司];Ealise T-SR正置荧光显微镜(日本尼康株式会社);1A1003型电子天平(上海越平科学仪器有限公司);SpectraMax MS型酶标仪(美国塞默飞世尔科技有限公司)。
1.2试药 腺苷、3-甲氧基苯甲醛、3-甲基苯甲醛、对甲基苯甲醛、3-氟苯甲醛、3-氯苯甲醛、3-溴苯甲醛、无水氯化锌、超干四氢呋喃、乙酸酐、丙酸酐、丁酸酐、异丁酸酐、甲醇钠、稀硫酸和无水吡啶,均购自安耐吉有限公司;甲苯、乙酸乙酯、石油醚和二氯甲烷,均购自国药集团化学试剂有限公司;无水甲醇(西陇科学股份有限公司);柱层析硅胶(烟台德信生物科技有限公司);薄层层析板GF254(烟台信德化学有限公司);生理盐水(石家庄四药有限公司);RPMI-1640培养基和磷酸盐缓冲液(PBS),均购自Gibco公司;胎牛血清、二甲基亚砜和氘代氯仿,均购自Sigma公司;乙二胺四乙酸(Amresco公司)。
1.3细胞 MCF-7乳腺癌细胞,由中国医学科学院细胞资源中心提供。
2 方法
2.1化合物的合成 合成路线见图1,部分方法参考文献[13-15]方法进行实验。
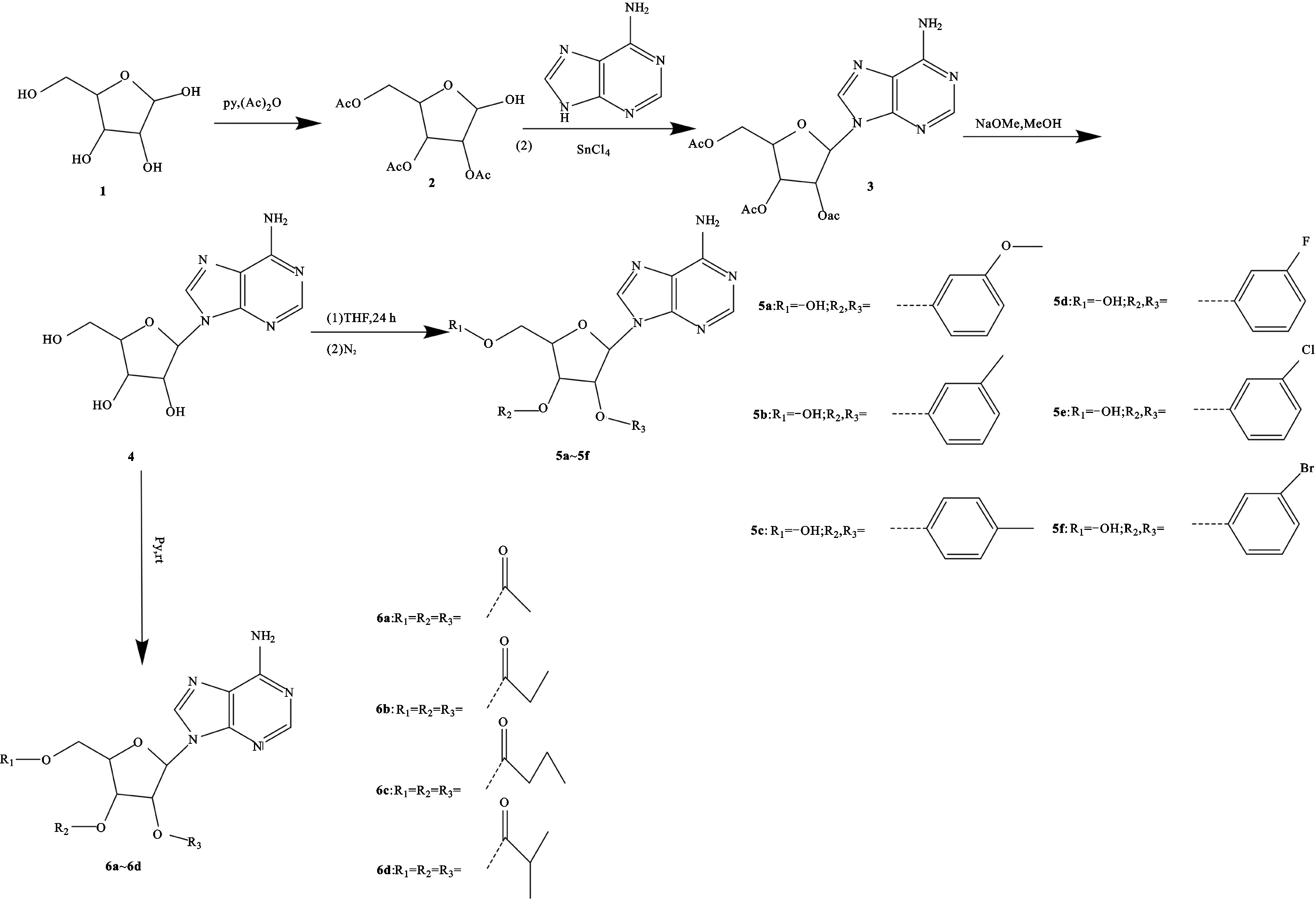
图1 目标化合物5a~5f和6a~6d的合成路线Fig.1 Synthesis route of target compounds 5a-5f and 6a-6d
化合物2即(2R,3R,4R,5R)-2-(乙酰氧基甲基)-5-羟基四氢呋喃-3,4-二基二乙酸酯的合成 取化合物1(1 g,0.003 7 mol),加入无水吡啶10 mL,再加入乙酸酐(2.24 mL,0.002 4 mol),室温下搅拌24 h后,薄层色谱检测反应完全,加入乙醇1 mL去除过量的乙酸酐,抽真空浓缩,用30 mL甲苯洗涤,重复3次,旋干呈黄色油状物,P2O5真空干燥得到化合物2(2.10 g,产率为99.0%),为白色粉末。1H NMR(400 MHz,DMSO-d6)δH:5.98(d,J=0.6 Hz,1H),5.25~5.16(m,2H),4.33~4.22(m,2H),4.06~3.97(m,1H),2.08~1.99(m,12H);MS(m/z):319.10[M+H]+。
化合物3即(2R,3R,4R,5R)-2-(乙酰氧基甲基)-5-(6-氨基-9H-嘌呤-9-基)四氢呋喃-3,4-二乙酸二酯的合成 在室温和氮气保护下,将化合物2(0.355 g,0.001 1 mol)和腺嘌呤(0.161 g,0.001 2 mol)加入到氯化锡(0.62 g,0.002 4 mol)中,再将混合物滴加到无水乙腈30 mL中,得到浅黄色溶液,搅拌18 h后,减压蒸馏。在冰浴搅拌条件下逐滴加入碳酸钠(0.69 g,0.006 5 mol),反应直到气泡停止,得到白色沉淀物。减压蒸馏后剩余白色粉末,将其用热氯仿重复萃取,浓缩,得到浅黄色油状物(0.539 g)。进行柱色谱分离(氯仿-甲醇=95∶5),得到化合物3(0.29 g,产率为67.1%),为无色油状物。1H NMR (400 MHz,DMSO-d6)δH:8.32(s,1H),8.12(s,1H),7.37(s,2H),6.16(d,J=5.5 Hz,1H),5.99(t,J=5.7 Hz,1H),5.58(dd,J=5.9,4.7 Hz,1H),4.41~4.33(m,1H),4.31(dd,J=5.1,3.8 Hz,1H),4.19(dd,J=11.7,5.5 Hz,1H),2.07(s,2H),1.98(d,J=11.1 Hz,7H);MS(m/z):384.13[M+H]+。
化合物4即(2R,3R,4R,5R)-2-(6-氨基-9H-嘌呤-9-基)-5-(羟甲基)四氢呋喃-3,4-二醇的合成 取化合物3(100 mg,0.000 25 mol),加入12.5 mL的甲醇钠溶液,用稀硫酸调节pH值至5.5。通过硅藻土过滤除去所得的白色沉淀物,并用无水甲醇洗涤;用硫酸镁干燥,旋转蒸发合并滤液和洗涤液,得到无色粉末(0.071 g,产率为100%)。从水中重结晶,在P2O5上真空干燥,得到化合物4(0.02 g,产率为33.3%),为无色针状物。1H NMR(400 MHz,DMSO-d6)δH:8.36(s,1H),8.14(s,1H),7.37(s,2H),5.88(d,J=6.2 Hz,1H),5.50~5.41(m,2H),5.21(d,J=4.5 Hz,1H),4.62(td,J=6.3,4.9 Hz,1H),4.15(td,J=4.8,2.9 Hz,1H),3.97(q,J=3.4 Hz,1H),3.68(dt,J=12.1,4.1 Hz,1H),3.56(ddd,J=11.7,7.3,3.6 Hz,1H);MS(m/z):268.10[M+H]+。
化合物5a即2′,3′-O-p-甲氧基苄叉腺苷(6-(6-氨基-9H-嘌呤-9-基)-2-(3-甲氧基苯基)四氢呋喃[3,4-d][1,3]二噁酚-4-基)甲醇的合成 取化合物4(1 g,0.003 7 mol),加入2 mL的超干四氢呋喃,加入无水氯化锌(3.5 g,0.257 mol),4 mL 3′-甲氧基苯甲醛,常温搅拌24 h后,薄层色谱法检测反应完全,停止反应。加入20 mL乙酸乙酯,滴加6 mL碳酸氢钠饱和溶液后过滤。用乙酸乙酯30 mL提取滤液中的有机相,重复3次,再用100 mL水洗涤有机相。抽真空浓缩得淡黄色油状物,进行柱色谱分离(50%~100%,乙酸乙酯/石油醚),得到白色固体5a(1.16 g,产率为80.2%)。1H NMR(400 MHz,DMSO-d6)δH:8.38(d,J=2.6 Hz,1H),8.17(d,J=2.2 Hz,1H),7.37(q,J=9.7,8.8 Hz,4H),7.16~7.10(m,1H),7.03(dd,J=19.7,9.7 Hz,1H),6.29(dd,J=4.9,3.0 Hz,1H),6.01(s,1H),5.52~5.47(m,1H),5.09(d,J=6.3 Hz,1H),4.36(t,J=3.8 Hz,1H),3.64~3.52(m,3H),1.90(s,3H);相对分子质量为385.38,MS(m/z):386.15[M+H]+。
化合物5b即(6-(6-氨基-9H-嘌呤-9-基)-2-(间甲苯基)四氢呋喃并[3,4-d][1,3]二氧代-4-基)甲醇的合成 取化合物4(1 g,0.003 7 mol),加入2 mL的超干四氢呋喃,加入无水氯化锌(3.5 g,0.257 mol),4 mL 3′-甲基苯甲醛。常温搅拌24 h后,薄层色谱法检测反应完全,停止反应。加入20 mL乙酸乙酯,滴加6 mL碳酸氢钠饱和溶液,过滤。用乙酸乙酯30 mL提取滤液中的有机相,重复3次,再用100 mL水洗涤有机相。抽真空浓缩得淡黄色油状物,进行柱色谱分离(50%~100%,乙酸乙酯/石油醚);得到白色固体5b(0.75 g,产率为54.3%)。1H NMR(400 MHz,DMSO-d6)δH:8.35(d,J=2.6 Hz,1H),8.13(d,J=2.2 Hz,1H),7.31(m,4H),6.26(dd,J=19.7,9.7 Hz,1H),5.96(s,1H),5.46(dd,J=4.9,3.0 Hz,1H),5.28(m,2H),5.04(dd,J=6.3,3.3 Hz 1H),4.33(m,1H),3.58(m,3H),1.70(s,3H);MS(m/z):370.17 [M+H]+。
化合物5c即(6-(6-氨基-9H-嘌呤-9-基)-2-(对甲苯基)四氢呋喃[3,4-d][1,3]二氧醇-4-基)甲醇的合成 取化合物4(1 g,0.003 7 mol),加入2 mL的超干四氢呋喃,加入无水氯化锌(3.5 g,0.257 mol),4 mL对甲基苯甲醛,常温搅拌24 h后,薄层色谱法检测反应完全,停止反应。加入20 mL乙酸乙酯,滴加6 mL碳酸氢钠饱和溶液,过滤。用乙酸乙酯30 mL提取滤液中的有机相,重复3次,再用100 mL水洗有机相。抽真空浓缩得淡黄色油状物,进行柱色谱分离(50%~100%,乙酸乙酯/石油醚);得到白色固体5c(0.95 g,产率为65.9%)。1H NMR(400 MHz,DMSO-d6) δH: 8.38(d,J=2.6 Hz,1H),8.27(d,J=2.2 Hz,1H),7.37(q,J=9.7,8.8 Hz,4H),7.31~7.25(m,1H),6.37(dd,J=19.7,9.7 Hz,1H),6.29(dd,J=4.9,3.0 Hz,1H),6.01(s,1H),5.52~5.47(m,1H),5.07 (dd,J=6.5,2.3 Hz,1H),4.36(t,J=3.8 Hz,1H),3.64~3.52(m,3H),2.0(s,3H);MS(m/z):370.14[M+H]+。
化合物5d即(6-(6-氨基-9H-嘌呤-9-基)-2-(3-氟苯基)四氢呋喃[3,4-d][1,3]二噁酚-4-基)甲醇的合成 取将化合物4(1 g,0.003 7 mol),加入2 mL的超干四氢呋喃,加入无水氯化锌(3.5 g,0.257 mol),4 mL 3′-氟苯甲醛,常温搅拌24 h后,薄层色谱法检测反应已完全,停止反应。加入20 mL乙酸乙酯,滴加6 mL碳酸氢钠饱和溶液,过滤。用乙酸乙酯30 mL提取滤液中的有机相,重复3次,再用100 mL水洗有机相。抽真空浓缩得淡黄色油状物,进行柱色谱分离(50%~100%,乙酸乙酯/石油醚);得到白色固体5d(1.25 g,产率为89.4%)。1H NMR(400 MHz,DMSO-d6)δH:8.40 (s,1H),8.16 (s,1H),7.61~7.40(m,4H),7.47~7.35(m,1H),6.30 (d,J=2.8 Hz,1H),6.01(s,1H),5.50 (dd,J=6.5,2.8 Hz,1H),5.32(t,J=5.5 Hz,1H),5.07 (dd,J=6.5,2.3 Hz,1H),4.34 (dt,J=8.7,5.2 Hz,3H),3.56(t,J=3.8 Hz,1H),3.51(m,3H);MS(m/z):374.12[M+H]+。
化合物5e即(6-(6-氨基-9H-嘌呤-9-基)-2-(3-氯苯基)四氢呋喃并[3,4-d][1,3]二氧代-4-基)甲醇的合成 取化合物4(1 g,0.003 7 mol),加入2 mL的超干四氢呋喃,加入无水氯化锌(3.5 g,0.257 mol),4 mL 3′-氯苯甲醛,常温搅拌24 h后,薄层色谱法检测反应已完全,停止反应。加入20 mL乙酸乙酯,滴加6 mL碳酸氢钠饱和溶液,过滤。用乙酸乙酯30 mL提取滤液中的有机相,重复3次,再用100 mL水洗有机相。抽真空浓缩得淡黄色油状物,进行柱色谱分离(50%~100%,乙酸乙酯/石油醚);得到白色固体5e(1.15 g,产率为78.5%)。1H NMR(400 MHz,DMSO-d6)δH: 8.34 (s,1H),8.13 (s,1H),7.61(q,J=9.7,8.8 Hz,3H),7.52~7.47(m,1H),7.37(dd,J=19.7,9.7 Hz,1H),6.19(dd,J=4.9,3.0 Hz,1H),6.01(s,1H),5.51 (dd,J=6.5,2.8 Hz,1H),5.21(s,1H),5.08 (dd,J=6.6,2.4 Hz,1H),4.34 (td,J=5.1,2.4 Hz,1H),3.52 (tt,J=11.5,6.4 Hz,2H);MS(m/z):390.09[M+H]+。
化合物5f即(6-(6-氨基-9H-嘌呤-9-基)-2-(3-溴苯基)四氢呋喃[3,4-d][1,3]二噁酚-4-基)甲醇的合成 取化合物4(1 g,0.003 7 mol),加入2 mL的超干THF,加入无水氯化锌(3.5 g,0.257 mol),4 mL 3′-溴苯甲醛,常温搅拌24 h后,薄层色谱法检测反应已完全,停止反应。加入20 mL乙酸乙酯,滴加6 mL碳酸氢钠饱和溶液,过滤。用乙酸乙酯30 mL提取滤液中的有机相,重复3次,用100 mL水洗有机相。抽真空浓缩得淡黄色油状物,进行柱色谱分离(50%~100%,乙酸乙酯/石油醚);得到白色固体5f(0.98 g,产率为60.4%)。1H NMR(400 MHz,DMSO-d6)δH:8.51(s,1H),8.24(s,1H),7.37(m,6H),6.30(d,J=4.9,3.0 Hz,1H),6.01(s,1H),5.52~5.47(m,1H),5.08(m,1H),4.40(t,J=3.8 Hz,1H),3.56(m,2H);MS(m/z):434.02[M+H]+。
化合物6a即(2R,3R,4R,5R)-2-(乙酰氧基甲基)-5-(6-氨基-9H-嘌呤-9-基)四氢呋喃-3,4-二乙酸二酯的合成 取化合物4(1 g,0.003 7 mol),加入10 mL无水吡啶,再加入1.12 mL乙酸酐,搅拌16 h后,薄层色谱法检测反应已完全,加入冰冷的无水乙醇,停止反应。抽真空浓缩得淡黄色油状物,进行柱色谱分离(50%~100%,乙酸乙酯/石油醚);得白色固体6a(1.46 g,产率为99.1%)。1H NMR(400 MHz,DMSO-d6)δH:8.32(s,1H),8.12(s,1H),7.37(s,2H),6.16(d,J=5.5 Hz,1H),5.99(t,J=5.7 Hz,1H),5.58(dd,J=5.9,4.7 Hz,1H),4.41~4.33(m,1H),4.31(dd,J=5.1,3.8 Hz,1H),4.19(dd,J=11.7,5.5 Hz,1H),2.07(s,2H),1.98(d,J=11.1 Hz,7H);MS(m/z):394.14[M+H]+。
化合物6b即(2R,3R,4R,5R)-2-(6-氨基-9H-嘌呤-9-基)-5-((丙酰氧基)甲基)四氢呋喃-3,4-二丙酸二酯的合成 取化合物4(1 g,0.003 7 mol),加入10 mL无水吡啶,再加入1.53 mL丙酸酐,搅拌6 h后,薄层色谱法检测反应已完全,加入冰冷的无水乙醇,停止反应。抽真空浓缩得淡黄色油状物,进行柱色谱分离(50%~100%,乙酸乙酯/石油醚);得白色固体6b(1.47 g,产率为90.0%)。1H NMR(400 MHz,DMSO-d6) δH:8.35 (s,1H),8.17 (s,1H),7.43 (s,2H),6.20 (d,J=5.3 Hz,1H),6.04 (t,J=5.6 Hz,1H),5.66 (t,J=5.4 Hz,1H),4.42 (dd,J=11.7,3.7 Hz,1H),4.36 (q,J=4.6 Hz,1H),4.26 (dd,J=11.8,5.3 Hz,1H),2.45~2.38 (m,2H),2.35~2.28(m,4H),1.07 (t,J=7.5 Hz,3H),0.99(td,J=7.4,5.7 Hz,6H);MS(m/z):436.19[M+H]+。
化合物6c即(2R,3R,4R,5R)-2-(6-氨基-9H-嘌呤基)-5-(丁酰氧基)甲基四氢呋喃-3,4-二丁酸酯的合成 取化合物4(1 g,0.003 7 mol),加入10 mL无水吡啶,再加入1.94 mL丁酸酐,搅拌6 h后,薄层色谱法检测反应已完全,加入冰冷的无水乙醇,停止反应。抽真空浓缩得淡黄色油状物,进行柱色谱分离 (30%~100%,乙酸乙酯/石油醚);得白色固体6c(1.47 g,产率为82.1%)。1H NMR(400 MHz,DMSO-d6)δH:8.31 (s,1H),8.12 (s,1H),7.37(s,2H),6.16 (d,J=5.3 Hz,1H),6.02 (t,J=5.6 Hz,1H),5.67~5.60(m,1H),4.38 (dd,J=11.8,3.7 Hz,1H),4.32(q,J=4.7 Hz,1H),4.22 (dd,J=11.7,5.2 Hz,1H),2.43~2.15 (m,5H),1.56 (d,J=7.3 Hz,1H),1.56~1.48(m,1H),1.46 (qd,J=7.3,5.6 Hz,4H),1.20(s,1H),0.89 (d,J=7.4 Hz,2H),0.85 (s,1H),0.88~0.74 (m,6H);MS(m/z):478.23[M+H]+。
化合物6d即(2R,3R,4R,5R)-2-(6-氨基-9H-嘌呤基)-5-(异丁酰氧基)甲基四氢呋喃-3,4-二基双(异丁酸)的合成 取化合物4(1 g,0.003 7 mol),加入10 mL无水吡啶,再加入1.96 mL异丁酸酐,搅拌6 h后,薄层色谱法检测反应已完全,加入冰冷的无水乙醇,停止反应。抽真空浓缩得淡黄色油状物,进行柱色谱分离(30%~100%,乙酸乙酯/石油醚);得白色固体6d(1.43 g,产率为80.0%)。1H NMR(400 MHz,DMSO-d6) δH:8.33 (s,1H),8.15 (s,1H),7.38 (s,2H),6.20 (d,J=5.0 Hz,1H),6.02 (t,J=5.4 Hz,1H),5.68 (t,J=5.3 Hz,1H),4.41~4.36 (m,2H),4.28 (dd,J=13.2,6.2 Hz,1H),2.62 (dt,J=14.1,7.1 Hz,1H),2.57~2.53 (m,1H),1.14 (d,J=7.0 Hz,7H),1.05 (ddd,J=13.4,7.0,4.2 Hz,14H);MS(m/z):478.32[M+H]+。
2.2抗肿瘤细胞增殖活性实验 细胞培养:以RPMI-1640(含体积分数为10%的胎牛血清,10 g·L-1双抗)为培养基,将传代至少3次以上、处于对数生长期的MCF-7细胞种板培养,具体种板条件为:96孔板,细胞密度为1×104个·孔-1,将培养板置于温度为37 ℃、体积分数为5%CO2的恒温细胞培养箱中培养24 h,沿孔板底部边缘处轻轻吸除培养板中培养基,待给药[16]。
给药:细胞培养板中每孔加入100 μL含有不同浓度药物的细胞培养基,其中阴性对照组为RPMI-1640培养基(含体积分数为10%的胎牛血清,10 g·L-1双抗),阳性对照组为含5 g·L-1DMSO的生理盐水,置于温度为37 ℃、体积分数为5%CO2的恒温细胞培养箱中培养24 h,在显微镜下观察细胞形态并拍照[17]。
细胞染色及细胞活性测试:将给药后24 h的培养板取出,每孔分别加入10 μL MTT染色剂,轻轻振摇,在37 ℃、体积分数为5%CO2的恒温细胞培养箱中孵育4 h;取出后,吸出培养板中上层清液,加入100 μL DMSO,置于摇床中,转速调至110 r·min-1,时间为15 min。用酶标仪检测490 nm波长处的吸光度值A,计算各样品的细胞活性,细胞活性=样品吸光度值A÷阴性对照组吸光度值A×100%,每个样品做3个复孔。
2.3分子对接 利用PYMOL2.3.4软件对受体蛋白(PDB Code:4CFF)进行去水、去配体等操作,采用AutoDockTools软件对受体蛋白进行加氢、平衡电荷等修饰,用Grid程序下的Grid Box命令打开Grid Option工具对受体蛋白进行处理,配体结合口袋的大小由各个方向上格点的数量和格点间距共同决定,因此调整蛋白每个方向上格点的数量,结合口袋的中心以及格点的间距,先将格点间距设为1,再调整结合口袋体积使预对接的分子在其最伸展的状态下也能在盒子内转动,口袋中心即设定为结合位点中心,并将受体蛋白和配体小分子分别转化为pdbqt格式[18]。
3 结果与讨论
3.1化合物的合成分析 所有化合物都经过核磁共振氢谱、质谱分析确证结构。
在合成路线设计上,化合物5a~5f需在糖环2′ 和3′ 羟基位置共成环以引入亲脂性基团。考虑到5′羟基和氨基的反应活性与2′ 和3′ 羟基接近,在研究初期,尝试了采用叔丁氧羰基(BOC)、叔丁基二甲基硅醚(TBDMS)、苄基(Bn)等基团首先对5′ 羟基和氨基进行临时性保护,结果发现,这些基团对氨基和羟基的选择性差,副产物多,保护效率低。而最终采用图1中的合成路线,不需要保护5′ 羟基和游离的氨基,能特异性修饰2′和3′羟基位点,并且产率较高。
此外,在糖环2′和3′羟基共成环的合成条件选择上,即从化合物4制备5a~5f的过程中,为了提高反应效率,以5a为例,针对合成条件进行了筛选,实验结果显示,无氮气保护反应12、24、36 h,产率分别为33.3%、25.5%、18.7%。进一步控制反应时间并尝试氮气保护条件,实验结果显示,氮气保护有利于提高反应收率,这可能是因为减少了醛基在反应过程中的氧化副产物。此外,在氮气保护下,不同反应时长也显示了不同的产率,反应12、24、36 h时,产率分别为45.6%、80.2%、67.3%。最终,化合物5a的反应条件选择使用氮气保护,反应24 h。
3.2抗肿瘤细胞增殖活性实验分析 实验采用MCF-7肿瘤细胞的体外抗增殖模型,测试了化合物的抗肿瘤活性,实验结果显示,化合物4、5a、5b、5c、5d、5e、5f、6a、6b、6c和6d对MCF-7细胞的IC50分别为0.52、0.17、0.40、7.13、0.45、0.14、2.6、2.6、481.34、152.84、62.17。2′、3′羟基取代成环的化合物(5a~5f)活性普遍强于糖环羟基全酯化取代的化合物(6a~6d),说明糖环5′羟基对抗肿瘤活性具有重要作用,可能与参与体内的磷酸化进程有关,不宜进行取代性修饰。
化合物5a和5e对MCF-7细胞的IC50分别为0.17、0.14 mmol·L-1,优于阳性对照(腺苷,化合物4),化合物5bIC50为0.40 mmol·L-1,与阳性对照相当。初步的构效关系提示:(1)引入的芳香环结构,苯环间位甲基取代化合物(5b,IC50=0.40 mmol·L-1)抗肿瘤活性显著优于对位(5c,IC50=7.13 mmol·L-1)。(2)芳环间位卤素原子取代,氯原子最佳,而当为溴原子时活性有所下降(5f,IC50=2.6 mmol·L-1),可能是溴原子半径较大,影响了分子与酶的活性中心的相互作用。
化合物的亲水性、疏水性是一个重要的理化性质,一般认为油水分配系数在-2~5之内才可能具有成药性,油水分配系数为0~2最佳。腺苷的lgP值偏低,经ChemDraw软件计算为-1.17,而研究设计的10个目标化合物的lgP值分别为0.73、1.35、1.35、1.02、1.42、2.6、-1.11、0.85、2.1、1.99,其中5a、5b、5c、5d、5e和6c的lgP值为0~2,均具有良好的亲水性、疏水性。结合抗肿瘤细胞实验结果,目标化合物抗肿瘤活性增强,可能与改善亲脂性而使化合物更容易穿透细胞膜有关。研究结果提示,采用糖环2′ 和3′ 羟基共修饰策略来增加糖环上的亲脂性可能是提高腺苷类似物抗肿瘤活性的有效修饰方式。
一些研究报道了不同策略对腺苷其他位点进行的修饰。如Parker W B等[19]将2′ 羟基换成了F原子,保留碱基上有游离氨基和糖环上3′ 和5′ 羟基,抗肿瘤活性增强。如刘连等[8]去掉腺苷碱基3′ 位的N,保留游离氨基,糖环替换成其他苯环取代基,也提高了抗肿瘤效果。这些修饰策略与本研究增加亲脂性的思路相互印证,提高腺苷分子亲脂性是提高抗肿瘤活性的重要因素,此外,本研究的修饰策略和这些工作可补充结合,有助于设计更多化合物。
3.3分子对接分析 AutoDock Vina 是由Scripps实验室开发的开源分子对接软件,该软件采用拟牛顿方法进行局部优化,打分函数结合了基于经验打分和知识打分函数的优点,通过计算受体-配体复合物的空间效果、排斥作用、氢键、疏水相互作用以及分子的灵活性等值综合打分,评估其亲和力,作为衡量配体是否能与受体分子有效结合的重要指标[20-21]。
以从PDB数据库获得的AMPK蛋白(PDB Code:4CFF)[20]为受体,将活性最好的化合物5e与AMPK受体蛋白进行分子对接分析,对接区域限定为F链中的结合口袋区,见图2。结果显示,5e与受体的结合能为-8.6 kcal·mol-1,高于阳性对照(化合物4,结合能为-6.3 kcal·mol-1),提示其较阳性对照,与靶蛋白相互作用更强。
进一步对5e最优构象与受体蛋白的结合模式进行了相互作用分析。由图2A可知,氨基酸残基Val96、Ser97与5e配体小分子糖环部位形成氢键相互作用,氨基酸残基Asn144、Glu143、Val24与5e配体小分子碱基部位形成氢键相互作用。由图2B可知,氨基酸残基Ala156、Met93作用与5e中2′ 和3′ 羟基共成环引入的苯环取代基形成疏水相互作用,这些作用共同促使了化合物5e与AMPK受体蛋白更好的结合。
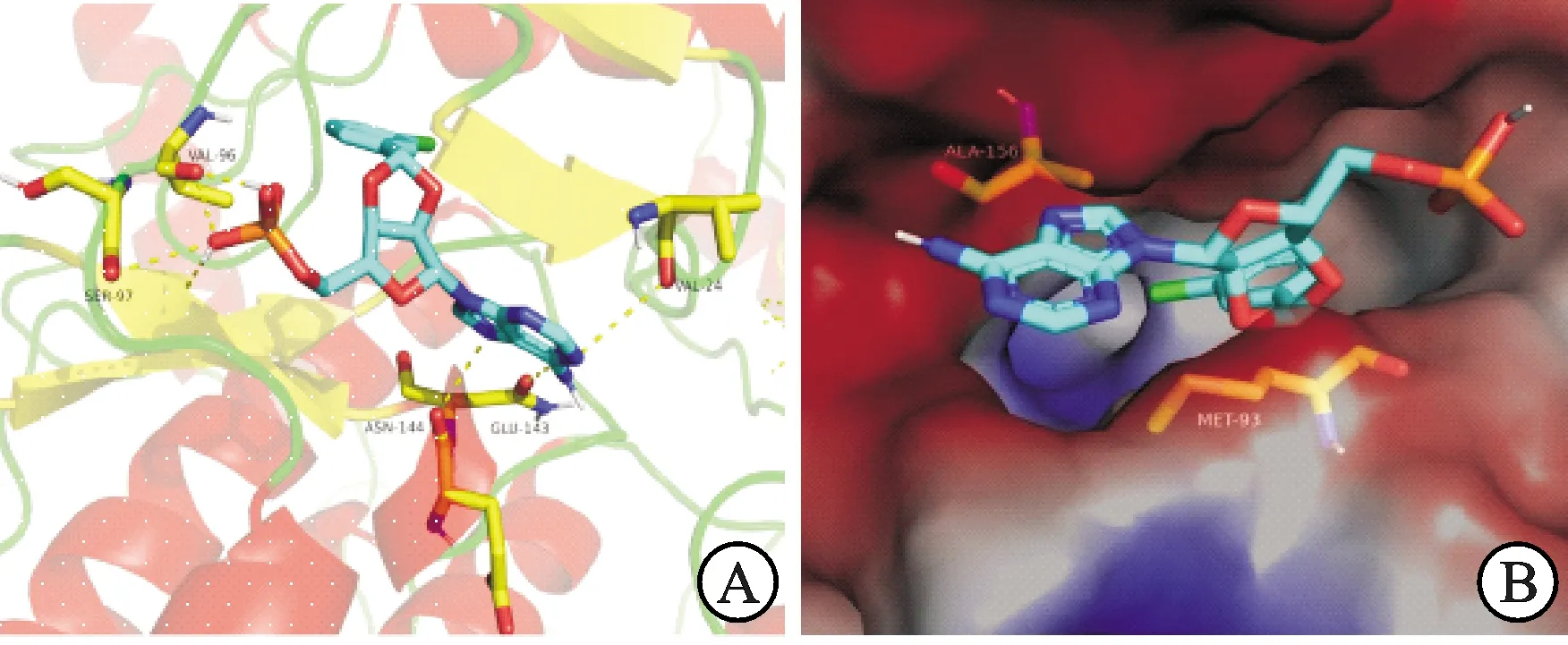
图2 5e与AMPK 酶的结合模式(A)以及与AMPK结合位点的表面形状(B)注:图A中蓝色分子为5e,黄线为氢键;图B中蓝色分子为5e,红色部分为亲水区域,蓝色部分为疏水区域。Fig.2 Binding pattern of 5e to AMPK enzyme(A) and surface shape of AMPK binding sites (B)Notes:in Figure A,the blue molecule is 5e,and the yellow line is hydrogen bond.In Figure B,the blue molecule is 5e,the red part is the hydrophilic region,and the blue part is the hydrophobic region.
4 结论
本文通过糖环2′和3′羟基成环以及羟基共成酯的修饰策略,合成了10个新结构腺苷衍生物,其结构经1H NMR和MS鉴定。体外抗肿瘤活性研究显示,大多化合物显示出了良好的抗肿瘤增殖活性,初步证实,采用糖环2′ 和3′ 羟基共取代成环策略来增加糖环上的亲脂性可能是提高腺苷类似物抗肿瘤活性的有效修饰方式。其中活性最好的化合物5e对MCF-7细胞的体外抗肿瘤活性IC50为0.14 mmol·L-1,优于阳性对照腺苷。利用分子对接技术显示目标化合物5e中2′ 和3′ 羟基共成环引入的苯环取代基增加了5e和AMPK的结合能。本研究可为以腺苷为母体的抗肿瘤化合物的设计和合成提供新的思路。
猜你喜欢
可再生能源(2022年5期)2022-06-09
城市道桥与防洪(2022年3期)2022-05-08
检察风云(2022年5期)2022-04-05
青少年科技博览(中学版)(2022年1期)2022-03-28
文萃报·周五版(2021年38期)2021-09-29
安全与环境工程(2021年2期)2021-04-02
读与写·下旬刊(2018年6期)2018-07-14
今日农药(2017年10期)2017-11-14
科学与财富(2017年17期)2017-06-16
中国医学创新(2017年11期)2017-05-10
