
高效液相色谱串联质谱法测定豆芽中4-氯苯氧乙酸、6-苄基腺嘌呤残留量的不确定度评定
2021-03-11 08:20唐韵熙
现代食品 2021年1期
◎ 唐韵熙
(重庆市食品药品检验检测研究院,重庆 401121)
豆芽因其产量高、价格便宜、有利于消化且加工方便,成为人们餐桌上的日常食品,受到普遍欢迎。但在豆芽生产过程中,各地多次出现了黑作坊生产的“毒豆芽”,严重危害着广大消费者的健康。其大多是不法商贩为了提高豆芽的产率,缩短豆芽生长周期,在豆芽生产过程中添加4-氯苯氧乙酸(4-CPA)、6-苄基腺嘌呤(6-BA)等植物生长调节剂以及抗生素类药物,促进豆芽下胚轴粗大,减少根部萌发,加速细胞分裂,且能起到抗菌、杀菌作用,使豆芽在生长过程中免受微生物的侵蚀。经常食用这些“毒豆芽”,会使人体内残留高剂量的药物,可能影响中枢神经[1-9]。《国家食品药品监督管理总局、农业部、国家卫生和计划生育委员会关于豆芽生产过程中禁止使用6-苄基腺嘌呤等物质的公告》(2015年第11号)[10]中明确规定,生产者不得在豆芽生产过程中使用4-氯苯氧乙酸钠、6-苄基腺嘌呤;豆芽经营者不得经营含4-氯苯氧乙酸钠、6-苄基腺嘌呤的豆芽。
植物生长调节剂的化学性质和生物特性因其种类繁多而各有异同,有较强的基质效应,残留量低,为提高检测质量与准确度,需对检测方法进行不确定度评定。目前,植物生长调节剂仅有6-苄基腺嘌呤、吲哚乙酸、吲哚丁酸和多效唑的非标准方法的不确定度报道[11-12],因此,本文建立了高效液相色谱-串联质谱法同时检测豆芽中4-CPA、6-BA 含量的测定方法,并且依据《测量不确定度评定与表示》(JJF 1059.1—2012)[13]、《化学分析中不确定度的评估指南》[14]的相关规定,对该方法的不确定度进行评定,找出影响测定结果的主要因素,并进行不确定度分析计算,进而对检测结果的准确性进行评判,提高检测质量。
1 材料与方法
1.1 仪器与试剂
AB5500 串联三重四极杆质谱联用仪(美国AB SCIEX 公司)、1290 高效液相色谱仪(美国安捷伦公司)、3-30KS 型冷冻离心机(德国Sigma 公司)、ML-886 涡旋仪(海门市其林贝尔仪器)、Turbovap LV 全自动浓缩仪(英国Biotage 公司)、Milli-Q reference 型纯水机(美国MILLIPORE 超纯水系统)、MSA225S-100-DU 电子分析天平(十万分之一,德国Sartorius 公司)、ME203/02 电子分析天平(千分之一,瑞士METTLER TOLEDO 公司)。
4-氯苯氧乙酸(4-CPA)标准品(德国Dr.Ehrenstorfer公司,纯度:98.11%)、6-苄基腺嘌呤(6-BA)标准品(德国Dr.Ehrenstorfer 公司,纯度:98.51%)、甲醇、乙腈、甲酸、无水硫酸镁、无水乙酸钠、Bondesil-C18。
1.2 标准溶液的配制
分别准确称取10.73 mg 4-CPA、10.24 mg 6-BA 于10 mL 容量瓶中,甲醇溶液定容,配制成浓度分别为1.053 mg·mL-1、1.009 mg·mL-1的标准储备液。
混合标准溶液1 配制[c(4-CPA)=10.53 μg·mL-1,c(6-BA)=10.09 μg·mL-1]:分别移取100 μL 4-CPA和100 μL 6-BA 标准储备液至10 mL 容量瓶,用甲醇定容至刻度。
混合标准溶液2 配制[c(4-CPA)=1.053 μg·mL-1,c(6-BA)=1.009 μg·mL-1]:准确移取1 mL 混合标准溶液1 至10 mL 容量瓶,用甲醇定容至刻度。
标准系列工作溶液配制:分别准确移取10 μL、20 μL、40 μL 混合标准 溶 液2,10 μL、15 μL、20 μL 混合标准溶液1 于1 mL 容量瓶中,用阴性基质定容至刻度。
1.3 方法
1.3.1 试样制备
称取5 g均质的豆芽样品于50 mL聚丙烯离心管中,加入10 mL 的1%甲酸乙腈溶液,振荡涡旋5 min,加入2 g 无水硫酸镁、0.5 g 无水乙酸钠,迅速振摇,于9 000 r·min-1离心10 min,准确移取4 mL 上清液至干净离心管中,45 ℃氮吹干,加入1 mL 甲醇溶解,超声5 min,转移上述溶解液于1.5 mL 净化小管中(内含75 mg 无水硫酸镁,25 mg C18),涡旋2 min,10 000 r·min-1离心5 min,取上清液过0.22 μm 滤膜,待LC/MS/MS 分析。
1.3.2 液相色谱
色谱柱:Acquity UPLC BEH C18(100 mm×2.1 mm,1.7 μm);柱温:40 ℃;流动相A:0.1 %甲酸水溶液;流动相B:乙腈。梯度洗脱程序:0 ~0.5 min,10% B,0.5 ~2.5 min,10% B →90% B,2.5 ~4.5 min,90% B,4.5 ~4.6 min,90% B →10% B,4.6 ~6.5 min,90% B,流速为0.3 mL·min-1,进样量1 μL。
1.3.3 质谱条件
电喷雾(ESI)离子源,负离子模式;扫描模式:多反应监测(MRM);离子源温度:550 ℃;离子源电压:4 500 V;气帘气压力:275.79 kPa;GAS1:379.21 kPa,GAS2:379.21 kPa;目标化合物的监测离子对,去簇电压(DP)和碰撞能(V)参数见表1。
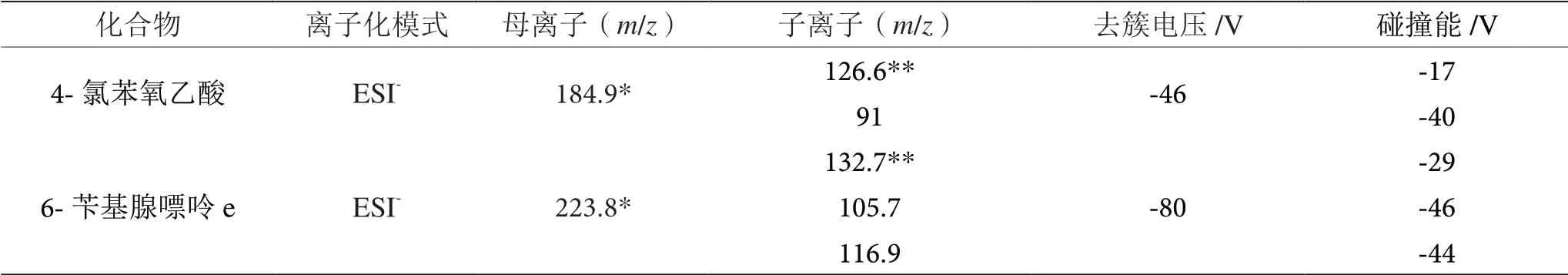
表1 4-CPA 及6-BA 的母离子、子离子及其他质谱参数表
1.3.4 数学模型
样品中待测组分的含量的计算公式为:

式中,X-样品中待测组分的含量,单位为μg·kg-1;C-从标准曲线得出的样品溶液中待测组分的浓度,单位为ng·mL-1;V-样品溶液最终定容体积,单位为mL;m-称样量;fr-待测组分的回收率(%);2.5-稀释倍数。
2 结果与分析
2.1 样品的不确定度来源
根据试验测试过程,分析影响豆芽试样中4-CPA及6-BA 残留量测定结果的各种不确定的因素,评价不确定度来源主要有:样品前处理制备、标准溶液的配制(包括储备液与混合中间溶液的配制过程)、标准工作溶液配制、根据拟合标准曲线求得样品浓度、回收率以及测试仪器。
2.2 由样品前处理引入的相对不确定度urel(样品)
样品前处理引入的不确定度主要有样品称量urel(m样)、加入提取试剂0.1%甲酸乙腈体积urel(V10mL)、移取4 mL提取试剂urel(V4mL),最终定容体积urel(V1mL),各分量不相关,具体见表2。合成样品前处理制备的相对不确定度为:
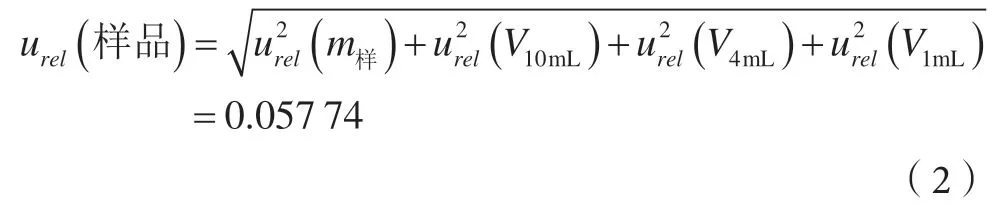
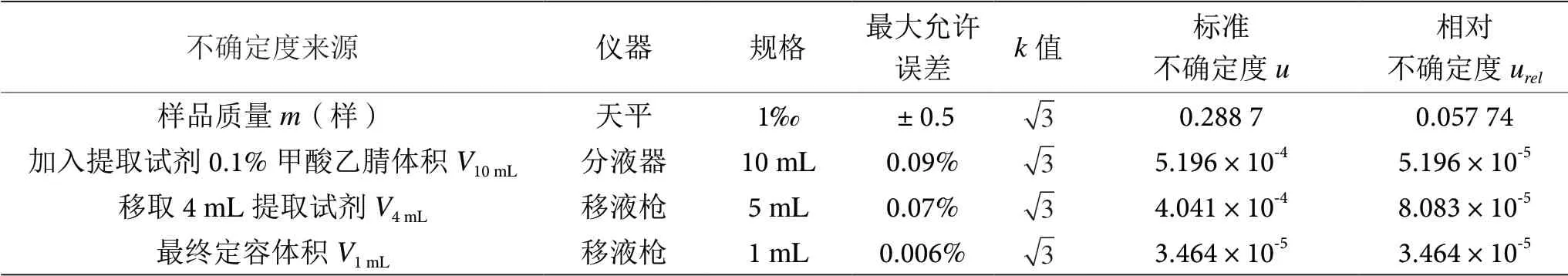
表2 样品前处理引入的不确定度表
2.3 标准溶液配制引入的相对不确定度urel(s)
标准溶液配制引入的相对不确定度主要来源于标准储备液配制urel(s1)、标准中间溶液配制urel(s2)和urel(s3)、标准工作溶液配制urel(s4)。
2.3.1 标准储备液配制引入的相对不确定度urel(s1)
其主要来源有:标准品纯度引入的不确定度urel(p)、标准品称量引入的不确定度urel(ms)、10 mL容量瓶校准和温度效应引入的不确定度urel(V10mL),具体见表3。

表3 标准储备液配制引入的相对不确定度表

续表3
2.3.2 混合标准中间溶液配制引入的相对不确定度urel(s2)和urel(s3)
混合标准中间溶液1、2 配制过程中引入的不确定度主要是:200 μL 移液枪、1 mL 移液枪、10 mL容量瓶以及容量瓶温度效应。根据移液枪校准证书,200 μL 和1 mL 移液枪最大允许误差分别是±0.3%和±0.4%,按矩形分布,k=,则两者产生的相对标准不确定度为:

由表3 可知,10 mL 容量瓶引入的相对不确定度urel(V10mL)=0.003 38,将上述分量合成标准不确定度为

2.3.3 标准工作溶液配制引入的相对不确定度urel(s4)
标准工作溶液是由豆芽阴性基质逐级稀释混合标准中间溶液配制而成的。配制过程中引入的不确定度分量及数值见表4[15],则合成各个标准溶液浓度点引入的相对不确定度为:

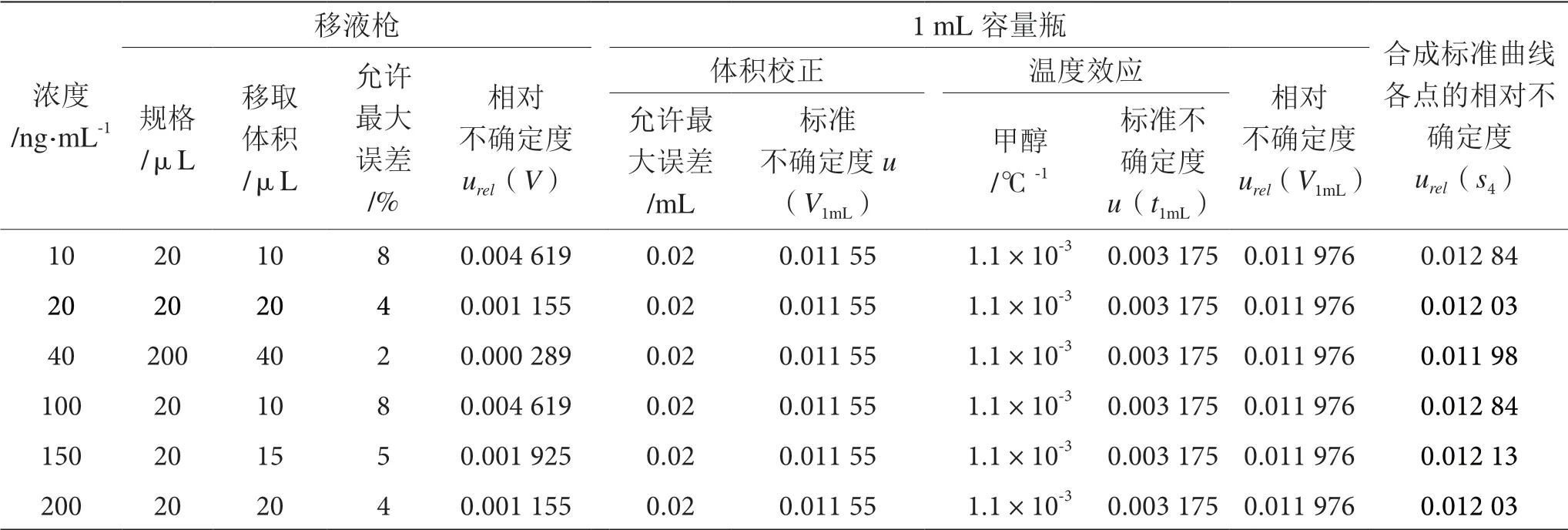
表4 标准工作溶液配制过程中引入的不确定度表
2.4 根据拟合标准曲线求得样品浓度引入的不确定度u(y)
将标准曲线的6 个浓度点每个进样3 次,以目标物的质量浓度为横坐标(C,ng·mL-1),目标物分子离子峰的峰面积为纵坐标(A)进行线性回归,回归方程、相关系数r见表5。
标准曲线拟合产生的不确定度按式(3)计算:
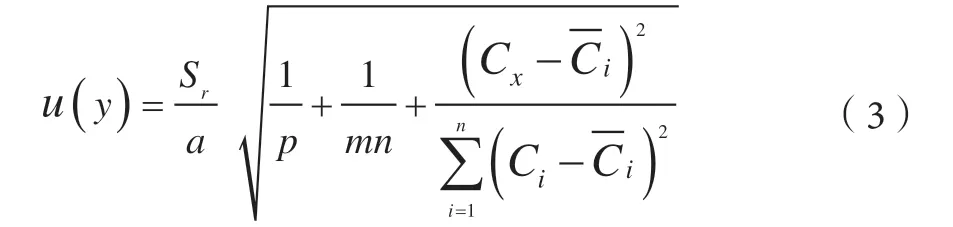
其中Sr按公式(4)计算:

式中Sr为标准溶液峰面积残差的标准差,Sr(4-CPA)=21.414 4;Sr(6-BA)=15.778 1;m为 标准溶液浓度点个数,m=6;n为标准溶液测定次数,n=3;p为待测物质测定次数,p=3,为标准溶液的平均浓度;a为斜率,b为截距。
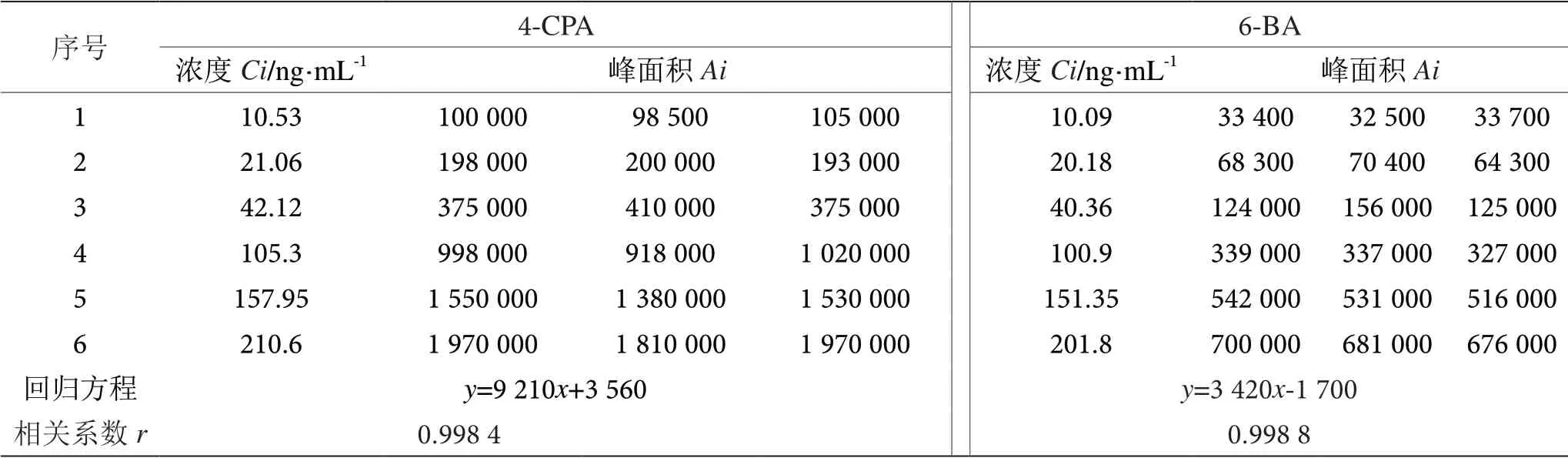
表5 标准曲线回归方程及线性表
将被测样品平行测定3 次,分别从标准曲线算出的4-CPA 和6-BA 化合质量浓度Cx(ng·mL-1)见表6。
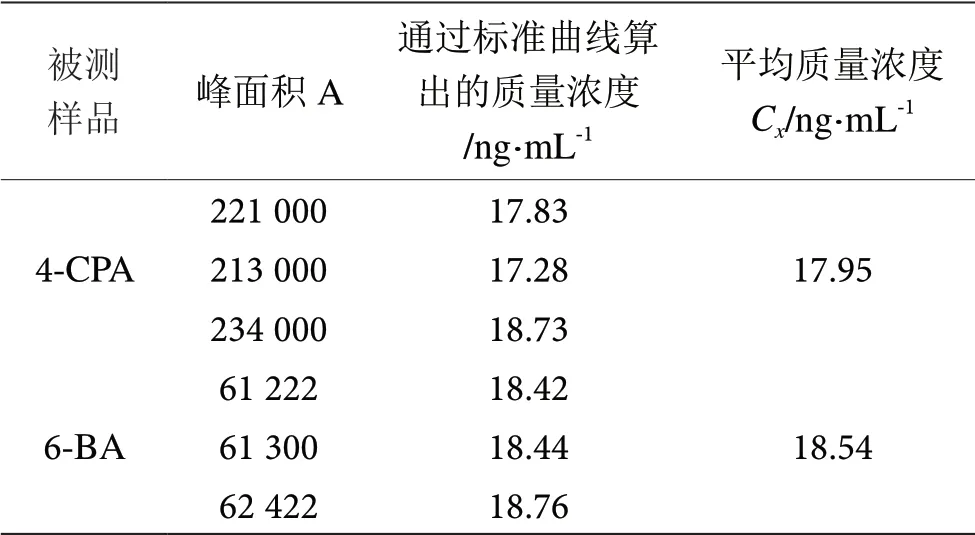
表6 被测样品的测定数据表
被测样品由标准曲线拟合产生的相对标准不确定度为:urel(y4-CPA)=0.041 56,urel(y6-BA)=0.040 13。
2.5 回收率测试过程引入的不确定度urel(fr)
在6份空白豆芽试样中分别加入绝对含量为52.65 ng的4-氯苯氧乙酸(4-CPA)和50.45 ng 6-苄基腺嘌呤(6-BA)混合标准溶液,按照1.3.1、1.3.2 和1.3.2 试样制备和仪器条件进行操作,结果见表7。按照式以下公式计算标准偏差、回收率不确定度和回收率相对不确定度。
计算标准偏差:
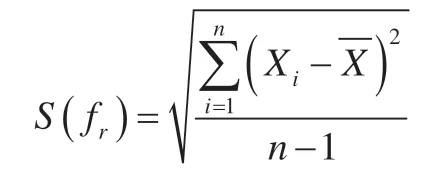
回收率不确定度:

回收率相对不确定度:
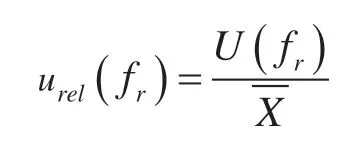
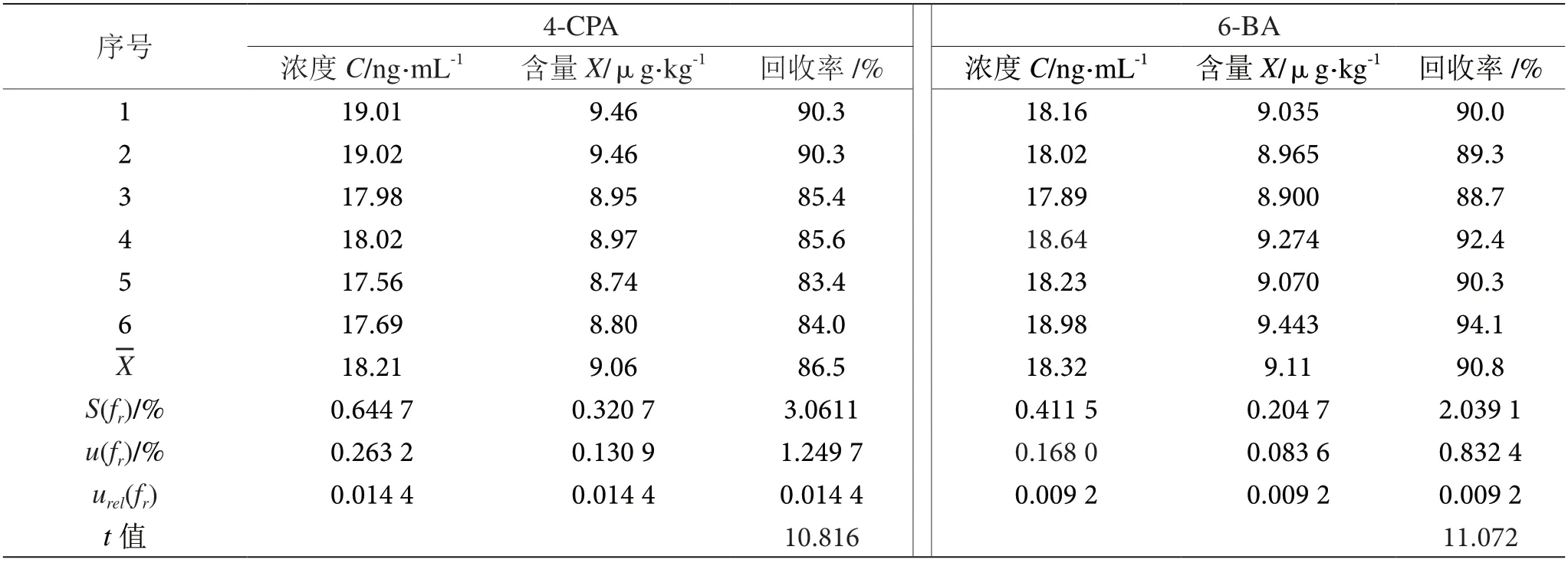
表7 回收率引入的不确定度表
用t检验法,,来检验回收率是否与100%存在显著差异性,结果见表7。由于t值>临界值(2.57),说明平均回收率与100%回收率之间有显著性差异,回收率矫正因子必须在计算公式中采用,以修正结果。
2.6 检测仪器引入的相对不确定度urel(fm)
根据《液相色谱-串联质谱 仪器校准证书》知,urel(fm)=4.0%。
3 标准不确定度的合成与扩展
3.1 各分量不确定度汇总
对于豆芽中4-CPA 和6-BA 测定结果有影响的各种不确定分量来源汇总见表8。

表8 各分量不确定度汇总表
3.2 合成标准不确定度
由于上述各因素的不确定度互不相关,合成豆芽中4-CPA 和6-BA 含量的相对不确定度为:
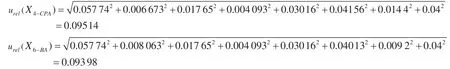
3.3 相对扩展不确定度
在95%置信水平下,k=2,4-CPA 的相对扩展不确定度为u(4-CPA)=9.06×0.095 14=0.86 μg·kg-1;6-BA的相对扩展不确定度为u(6-BA)=9.41×0.093 98=0.88 μg·kg-1。
3.4 测定结果
按照该方法测定豆芽中4-CPA 含量的结果为:X(4-CPA)=(9.06±0.86)μg·kg-1,k=2;X(6-BA)=(9.41±0.88)μg·kg-1,k=2。
4 结论
通过不确定度评定分析,采用超高效液相色谱-串联质谱法检测中豆芽中4-CPA 和6-BA 含量的过程中,样品前处理制备引起的不确定度最大,其次是由仪器以及标准溶液配制和通过标准曲线拟合求得样品浓度值过程带入。因此,在实际工作中应对样品前处理进行规范操作,同时选取精密性高的仪器进行检测,高精密度天平精密称量,高精密度的移液枪和容量瓶进行溶液配制,从而提高检测结果的准确度和可靠性。
猜你喜欢
农业工程学报(2022年5期)2022-06-22
世界科学技术-中医药现代化(2021年5期)2021-11-05
中老年保健(2021年4期)2021-08-22
基层中医药(2020年7期)2020-09-11
学苑创造·A版(2017年10期)2017-12-21
合成化学(2015年10期)2016-01-17
合成化学(2015年1期)2016-01-17
中国洗涤用品工业(2015年9期)2015-02-28
分析化学(2014年8期)2014-09-02
天然产物研究与开发(2014年6期)2014-04-27
