
以鸟苷为原料合成巴豆苷
2021-04-08 11:19孙莉萍焦黎明杨政楠潘心夏然陈磊山
精细石油化工 2021年2期
孙莉萍,焦黎明,杨政楠,潘心,夏然,陈磊山
(1.新乡学院生命科学与基础医学学院,河南 新乡 453003;2.新乡学院药学院,河南 新乡 453003)
巴豆苷(Crotonoside)又称异鸟苷(CAS号1818-71-9),是一种天然的核苷类化合物,最早从中药巴豆种子中分离得到。巴豆苷是巴豆重要的活性成分[1]。研究显示,巴豆苷具有多种生理活性,如能够增进肠道蠕动、降低血压,能够刺激大脑使AMP(腺苷酸)富集[2],对小鼠S180腹水瘤和Ehrlich实体肿瘤都有显著的抑制作用[3],可通过抑制FLT3和HDAC3/6在急性髓性白血病(AML)细胞中表现出选择性的后抑制作用[4],可以通过调节兔心室肌细胞钠钙通道而具有抗心律失常作用[5]。另外,巴豆苷是生物体内RNA的结构片段鸟苷的异构体,可以作为鸟苷的类似物嵌入RNA片段中并进行基因表达,从而在生化研究中具有重要用途[6]。
目前,巴豆苷除了从天然资源中分离得到以外,化学合成法主要有以下3种路线:1)2-取代(I、Cl或NH2)腺苷经过光照水解[7]或重氮化反应[8]得到,但是选择性差,收率很低,或者2-羟基-6-硫取代嘌呤核苷经氨解得到[9],但是原料合成难度大(图1,路线1);2)异鸟嘌呤经硅醚保护后和基团保护的核糖在TMSOTf或SnCl4催化下缩合,再脱除保护基得到,缺点是步骤多,收率低,异鸟嘌呤的合成难度也很大[10](图1,路线2);3)氨基咪唑甲酰胺核苷经环合仿生合成异鸟苷,但收率很低,不具有合成上的价值[11](图1,路线3)。以上3种反应路线限制了巴豆苷合成规模的扩大。从廉价易得的原料合成得到巴豆苷,可以避免从天然产物中分离、提取的繁琐步骤,扩大巴豆苷的来源;也可以解决现有化学合成方法中存在的收率低等问题。
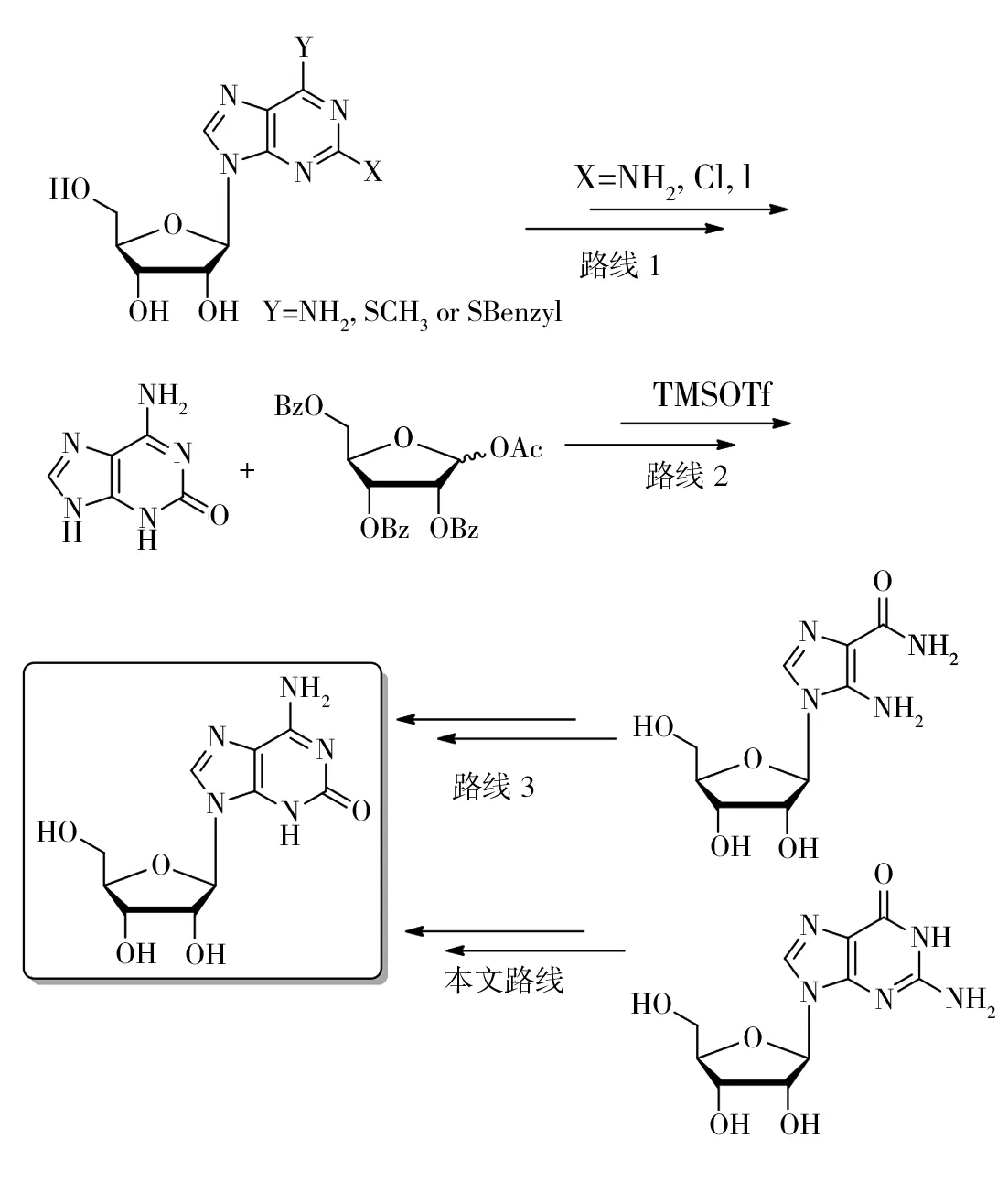
图1 巴豆苷合成路线比较
鸟苷是核苷化学领域的基础原料,可以通过发酵法规模化生产,价格低[12]。笔者以鸟苷原料,经过乙酰化、氯代、重氮化、氨解同时脱除乙酰基等4步,得到巴豆苷,总收率62%。合成路线见图2。

图2 巴豆苷的合成路线
1 实验部分
1.1 原料与仪器
鸟苷,分析纯,新乡拓新药业股份有限公司;其他所用试剂均为市售分析纯。
XT4型显微熔点测定仪,温度计未校正,北京科仪电光仪器厂;AV 400型核磁共振仪(DMSO-d6或CDCl3为溶剂,TMS为内标,德国Bruker公司。
1.2 合成方法
1.2.1 2′,3′,5′-三-O-乙酰基鸟苷(3)
鸟苷(2,2.83 g,10 mmol)加入到乙腈(100 mL)中,再加入DMAP(N,N-二甲基吡啶,0.18 g,0.5 mmol)和三乙胺(6.23 mL,45 mmol),搅拌30 min,冰水冷却,5 ℃下滴加醋酸酐(3.4 mL,36 mmol),滴加完毕,转移至油浴锅,缓慢升温至60 ℃,持续反应2 h,TLC显示原料反应完全,加入甲醇(5 mL),再继续反应30 min,冷却至室温,减压浓缩,有固体析出,过滤,水重结晶,得到白色固体0.388 g,收率95%。
白色固体,m.p. 231~233 ℃(文献[13]m.p. 231~233 ℃)。1H NMR(DMSO-d6, 400 MHz) δ 10.73(s, 1H), 7.94(s, 1H), 6.54(brs, 2H), 6.00(d,J=6.0 Hz, 1H), 5.80(t,J=5.6 Hz, 1H), 5.03(t,J=5.6 Hz, 1H), 4.40~4.24(m, 3H), 2.12(s, 3H), 2.05(s, 3H), 2.04(s, 3H);13C NMR(DMSO-d6, 100 MHz) δ 170.6, 169.96, 169.7, 157.1, 154.4, 151.6, 136.1, 117.3, 84.9, 80.0, 72.5, 70.8, 63.6, 21.0, 20.9, 20.7。
1.2.2 6-氯-2′,3′,5′-三-O-乙酰基鸟苷(4)
2′,3′,5′-三-O-乙酰基鸟苷(3,0.409 g,1 mmol)和N,N-二甲基苯胺(0.128 mL,1 mmol)溶于乙腈(10 mL),室温搅拌30 min,加入氯化苄基三乙铵(0.332 g,2 mmol),继续搅拌10 min,滴加POCl3(0.548 mL,6 mmol),升温至回流,持续反应2 h,TLC显示原料反应完全,冷却至室温,倒入冰水中,搅拌,用CH2Cl2(10 mL×3)萃取,有机相用饱和NaHCO3溶液(10 mL)洗涤,活性炭脱色和无水Na2SO4干燥,过滤、减压浓缩后加入异丙醇(5 mL),加热溶解,静置2 h,有固体析出,过滤,滤饼干燥后即得白色固体0.371 g,收率87%。
白色固体,m.p. 151~153 ℃(文献[13]m.p. 151~153 ℃)。1H NMR(CDCl3, 400 MHz) δ 7.88(s, 1H), 6.02~5.95(m, 2H), 5.75(d,J=4.4 Hz, 1H), 5.30(brs, 2H), 4.47~4.35(m, 3H), 2.15(s, 3H), 2.11 (s, 3H), 2.09(s, 3H);13C NMR(CDCl3, 100 MHz) δ 170.5, 169.6, 169.4, 159.1, 153.1, 151.9, 140.7, 125.8, 86.6, 80.0, 72.7, 70.5, 62.9, 20.7, 20.6, 20.4。
1.2.3 2-羟基6-氯-9-(2′,3′,5′-三-O-乙酰基)-β-D-呋喃核糖基嘌呤(5)
将4(0.428 g,1 mmol)加入到CHCl3(9 mL)中,搅拌溶解,加入H2O(1 mL),室温滴加亚硝酸异戊酯(0.403 mL,3 mmol),升温到60 ℃,继续反应2 h,TLC显示反应完全,减压浓缩,柱层析分离[流动相V(氯仿)∶V(甲醇)=9∶1],得到白色固体0.346 g,收率81%。
白色固体,m.p. 132~133 ℃(文献[14]m.p. 131~133℃)。1H NMR(CDCl3, 400 MHz) δ 10.80(s, 1H), 7.95(s, 1H), 6.18(d,J=5.6 Hz, 1H), 5.92(t,J=5.6 Hz, 1H), 5.66(t,J=4.8 Hz, 1H), 4.45~4.33(m, 3H), 2.12(s, 3H), 2.09(s, 3H), 2.06(s, 3H);13C NMR(CDCl3, 100 MHz) δ 170.3, 169.6, 169.4, 155.6, 153.0, 149.7, 138.9, 120.1, 86.3, 80.3, 73.2, 70.7, 63.1, 20.7, 20.5, 20.4。
1.2.4 巴豆苷(1)
将5(0.428 g,1 mmol)和饱和的NH3/CH3OH溶液(10 mL)加入到密封反应管中,加热到50 ℃反应12 h,冷至室温,释放压力,反应液真空浓缩,水重结晶,得到白色固体10.263 g,收率93%。
白色固体,m.p. 238~240 ℃(文献[15]m.p. 238~240 ℃)。1H NMR(DMSO-d6, 400 MHz) δ 10.88(s, 1H), 8.37(s, 1H), 7.83(brs, 2H), 5.81(d,J=6.0 Hz, 1H), 5.47(d,J=6.0 Hz, 1H), 5.19(d,J=4.8 Hz, 1H), 5.05(t,J=4.8 Hz, 1H), 4.52~4.48(m, 1H), 4.12~4.09(m, 1H), 3.93 (d,J=3.6 Hz, 1H), 3.67~3.52 (m, 2H);13C NMR(DMSO-d6, 100 MHz) δ 160.2, 156.0, 151.6, 137.1, 112.4, 84.1, 83.3, 75.6, 61.1。
2 结果与讨论
2.1 合成路线优化
合成得到中间体4之后,除了图2所示的路线,理论上也可以先氨解得到2-氨基腺苷(6),然后发生重氮化反应。但是,这条路线的收率只有32%,并且重氮化反应选择性差,还会有鸟苷(2)等副产物的产生。
2.1.1 2-氨基腺苷(6)合成
将化合物4(0.428 g,1 mmol)和饱和的NH3/CH3OH溶液(50 mL)加入到密封反应管中,加热到50 ℃反应12 h,冷至室温,释放压力,反应液真空浓缩,水重结晶,得到白色固体60.239 g,收率85%。
白色固体,m.p. 235~237 ℃(文献[15]m.p. 235~237 ℃)。1H NMR (DMSO-d6, 400 MHz) δ 7.93 (s, 1H), 6.79 (brs, 2H), 5.74 (brs, 2H), 5.73 (s, 1H), 5.45 (t,J=4.8 Hz, 1H), 5.37 (d,J=6.4 Hz, 1H), 5.10 (d,J=4.4 Hz, 1H), 4.55 (q,J=6.0 Hz, 1H), 4.13 (q,J=4.0 Hz, 1H), 3.93 (d,J=2.8 Hz, 1H), 3.68~3.64 (m, 1H), 3.58~3.52 (m, 1H);13C NMR (DMSO-d6, 100 MHz) δ 160.5, 156.7, 151.9, 136.8, 114.1, 87.6, 86.0, 73.7, 71.2, 62.2。
2.1.2 重氮化法合成巴豆苷(1)
化合物6(0.282 g,1 mmol)加入到H2O(10 mL)中,再加入NaNO2(0.24 g,3.5 mmol),搅拌溶解,滴加AcOH(0.27 mL,6.9 mmol),升温到50 ℃反应1 h,反应完毕,降至室温,用氢氧化钠溶液中和,减压浓缩,水重结晶,得到的固体柱层析分离,得到巴豆苷和鸟苷,收率分别为32%和21%。
鸟苷(2):白色固体,m.p. 238~240 ℃(文献[16]m.p. 238~240 ℃)。1H NMR (DMSO-d6, 400 MHz) δ 10.65 (s, 1H), 7.94 (brs, 1H), 6.45 (brs, 2H), 5.71 (d,J=6.4 Hz, 1H), 5.39 (d,J=6.0 Hz, 1H), 5.11 (d,J=4.8 Hz, 1H), 5.03 (t,J=5.2 Hz, 1H), 4.43 (q,J=5.6 Hz, 1H), 4.11 (q,J=4.4 Hz, 1H), 3.90 (q,J=4.4 Hz, 1H), 3.65-3.60 (m, 1H), 3.56~3.51 (m, 1H);13C NMR (DMSO-d6, 100 MHz) δ 157.2, 154.1, 151.8, 136.1, 117.2, 86.8, 85.7, 74.2, 70.8, 61.9。
2.2 2-羟基9-(2′,3′,5′-三-O-乙酰基)-β-D-呋喃核糖基嘌呤(5)合成条件优化
嘌呤环外氨基参与重氮化活性较低,其重氮化反应直接影响2位羟基的引入,是整条合成路线的关键步骤。对重氮化反应的关键条件重氮化试剂、溶剂、反应温度等进行了优化,结果见表1。结果表明,以水溶剂,以NaNO2/醋酸为重氮化试剂,在室温条件下反应4 h,无反应发生。提高温度至50 ℃,收率17%。溶剂中加入吡啶或氯仿作为混合溶剂,收率稍有提高,分别达到20%和33%。随后,对重氮化试剂进行了筛选。亚硝酸叔丁酯收率高于NaNO2/醋酸,6 mmol的亚硝酸叔丁酯可以得到60%的收率。当把亚硝酸叔丁酯改变为亚硝酸异戊酯时,收率提高到68%。随着温度的升高,收率提高,60 ℃时收率基本不再增加,达到72%。延长反应时间至6 h,收率78%。综上,合成6的优化条件为:6 mmol亚硝酸异戊酯,CHCl3/H2O为混合溶剂,60 ℃反应6 h,收率81%。

表1 2-羟基6-氯-9-(2′,3′,5′-三-O-乙酰基)-β-D-呋喃核糖基嘌呤(5)合成条件优化
2.3 关键反应步骤反应规模对反应的影响
为了增加该反应的实用性,考察了关键步骤氨解和重氮化的反应规模对收率的影响(表2)。结果显示,在适当延长反应时间后,氨解步骤可以扩大到200 g,重氮化步骤可以扩大到100 g,收率没有下降,分离纯化不需柱层析,操作及后处理均非常简便。

表2 不同反应规模对收率的影响
由表2可知,在反应规模达到200 g时,经过常规的分离提纯后,氨解收率可以达到88%。在反应规模达到100 g时,重氮化步骤的收率可以达到78%。对母液回收利用,收率还会有所增加,所以,总体上整条路线的收率高,分离纯化简便,具有一定的应用性。但是重氮化步骤尽管没有中间产物重氮类化合物的分离,降低了爆炸性危险系数,但是在反应规模扩大以后仍然具有一定的危险,这也是本方法的局限性所在。
3 结 论
以鸟苷为原料,用乙酰基保护糖环上的羟基,再和POCl3反应,将6位羰基转化为氯原子,继而在亚硝酸异戊酯作用下发生重氮化反应,将2位氨基转化为羟基,最后在饱和的NH3/CH3OH溶液中将6位氯原子氨解转化为氨基,同时脱除乙酰基,以4步和62%的总收率得到巴豆苷。该方法原料廉价易得,解决了传统方法中原料价格高的问题,产物成本低,高效、简便,易于扩大生产,反应规模可以扩大到200 g,收率基本没有降低,具有一定的应用前景。
猜你喜欢
湖北大学学报(自然科学版)(2020年3期)2020-06-18
饲料工业(2019年24期)2019-12-31
中国调味品(2018年7期)2018-07-17
物理化学学报(2017年8期)2017-12-18
婚姻与家庭·性情读本(2017年3期)2017-04-27
应用化工(2014年4期)2014-05-10
郑州大学学报(理学版)(2012年4期)2012-03-25
