
甘草泻心汤调控PERK-elF2α-CHOP信号通路保护应激态肠上皮细胞屏障的机制
2021-05-07 00:54钟继红倪思忆
中国药理学通报 2021年5期
沈 雁,钟继红,倪思忆,吕 宾
(1.浙江中医药大学附属第二医院消化内科,浙江 杭州 310005;2.浙江省中医院消化内科,浙江 杭州 310006)
溃疡性结肠炎(ulcerative colitis,UC)是一种累及结肠黏膜和黏膜下层的慢性复发性肠道炎症性疾病,其临床表现复杂多样,病理机制尚不十分明确。随着国民生活方式的日趋西化,我国UC发病率不断上升[1]。因此,深入探索UC的确切发病机制和寻找有效的治疗药物是迫切需要解决的临床实际问题。在UC的发病机制中,免疫紊乱是各种病因导致结肠炎症损伤的下游环节[2];而目前越来越多的研究证实,肠上皮细胞(intestinal epithelial cells,IECs)过度凋亡导致肠黏膜屏障损伤、局部免疫系统异常暴露是引起UC发病和病情进展的上游核心环节[3-4]。在这一环节中,内质网应激(endoplasmic reticulum stress,ERS)状态下激活的蛋白激酶R样内质网激酶(protein kinase R-like ER kinase,PERK)-真核细胞起始因子2α(eukaryotic initiation factor 2α,elF2α)-C/EBP同源蛋白(C/EBP homologous protein,CHOP)凋亡信号通路起到重要的中介作用[5]。大量证据表明,UC临床症状的消失并不代表长期治疗的成功,国际上已将“黏膜愈合”确立为UC的首要治疗终点和关键预后指标[6-7],保护肠黏膜屏障稳态、促进黏膜愈合已成为当前UC治疗药物开发的主要研究方向。甘草泻心汤(Gancao Xiexin Decoction,GXD)是起源于《伤寒论》的经典名方,可用于如反流性食管炎、复发性口腔溃疡、白塞氏病等多种黏膜损伤性疾病,国内众多学者的临床实践亦证实其对UC同样具有良好疗效[8-10],且长期使用未见明显不良反应。但该方的作用机制和作用靶点尚不清楚。本研究通过建立体外IECs的应激态模型,从GXD对细胞的活性、凋亡、通透性和细胞内PERK-elF2α-CHOP信号通路的影响方面阐明该方的部分作用机制。
1 材料
1.1 实验细胞系Caco-2细胞株购自美国ATCC公司,生长于含10%胎牛血清的DMEM高糖培养基中,置37 ℃、5%的CO2培养箱中常规培养,每1-2 d更换培养液。5-7 d后细胞生长达融合,按1 ∶2或1 ∶3传代。实验时取对数生长期细胞。
1.2 实验药物甘草泻心汤(炙甘草12 g、干姜9 g、半夏9 g、黄芩9 g、黄连3 g、党参9 g、大枣6 g),饮片购于浙江中医药大学附属第二医院中药房;按照成人-小鼠体表面积换算,确定小鼠临床甘草泻心汤等效剂量为8 g·kg-1·d-1;将上述饮片用蒸馏水浸泡,常规煎煮2次,混合过滤,浓缩为灌胃液后保存于-20 ℃冰箱。
1.3 试剂衣霉素(tunicamycin,Tm,美国Sigma-Aldrich公司,批号:654380);PERK、eIF2α、ATF4、CHOP一抗(美国Sigma-Aldrich公司,批号分别为:P0074、SAB4500729、SAB4300041、SAB4500631);羊抗兔和羊抗鼠二抗(杭州联科生物技术股份有限公司,批号分别为:GAR0072、GAM007);CCK-8试剂盒(上海联科生物公司);Annexin V-FITC/PI 凋亡试剂盒(美国Biouniquer公司)、PI染液(美国Sigma-Aldrich公司)、DMEM培养基、胎牛血清和胰蛋白酶(美国GIBCO公司);胶原酶Ⅺ、中性蛋白酶Ⅰ、山梨醇和DAPI液(美国Sigma-Aldrich公司)。
1.4 仪器3111型细胞培养箱、CO2培养箱、生物安全操作柜和离心机(美国Thermo公司);spectraMax Plus 384型全波长酶标仪(美国MD公司)、Accuri C6型流式细胞仪(美国BD公司)、Mini-Proten Tetra System电泳系统和ChemiDoc XRS+System凝胶成像仪(美国Bio-RAD公司)、Millicell-ERS跨膜电阻仪(美国Millipore公司)。
2 方法
2.1 含药血清的制备将30只BALB/c雄性小鼠随机分为两组:含药血清组给予甘草泻心汤8 g·kg-1·d-1,空白血清组给予等体积蒸馏水,每日2次,连续5 d(给药剂量=临床常用剂量×动物等效剂量系数×培养基内的稀释度)。采血前夜禁食不禁水,次日末次给药1 h后眼球摘除法取小鼠抗凝血,静置2 h后,3 000 r·min-1离心10 min,取血清56 ℃恒温水浴30 min灭活补体,0.22 μm除菌滤膜过滤除菌后,-80 ℃保存备用。
2.2 细胞分组造模Caco-2细胞分为正常对照(NC)组、模型(MC)组、GXD低剂量(GXD-L)、中剂量(GXD-M)和高剂量组(GXD-H),用Tm诱导法[7]建立细胞ERS模型。
2.3 CCK-8法检测细胞存活率细胞以5×107个·L-1的密度接种于96孔板,每孔200 μL,培养24 h,弃上清后分组给药:NC组为200 μL培养基中含有100 μL空白血清;MC组为200 μL培养基中含有100 μL空白血清和终浓度为10 mg·L-1的Tm;GXD-L、GXD-M和GXD-H组(分别含 5%、10%、20% 含药血清)为200 μL培养基中分别对应含有10、20和40 μL GXD含药血清,90、80和60 μL空白血清以及终浓度为10 mg·L-1的Tm。12 h药物干预结束后,PBS洗涤,每孔加入10 μL CCK-8工作液,37 ℃孵育2 h,450 nm读板,记录OD值。每组设6个复孔,同时设溶剂对照组。
2.4 流式细胞术检测细胞凋亡率和细胞周期调整细胞浓度为1×108个·L-1,3 mL接种于中号培养皿,培养24 h,弃上清后按上述分组和浓度比例给药,继续作用12 h后,胰酶消化细胞,PBS漂洗2次,2 000 r·min-1离心5 min。先用Binding buffer 悬浮细胞后加入5 μL Annexin V-FITC混匀,室温避光反应5-15 min,上机检测凋亡率;再加入70%冷乙醇固定12 h,加入1 mL PI染液,室温避光孵育20 min后,上机检测细胞周期。激发波长为488 nm,并用相应软件进行数据分析。每组重复3次。
2.5 Millicell-ERS电阻仪检测细胞跨膜电阻抗(TEER)调整细胞浓度为2×107个·L-1,200 μL/孔接种于Transwell顶室。底室中加入培养基600 μL。12 h细胞贴壁完全后,按上述分组和浓度比例给药,继续作用12 h后,用Millicell电阻仪检测单层膜细胞的TEER。每组设3个复孔。
2.6 FITC-dextran法检测细胞旁通透性按“2.5”中方法处理细胞并给药,48 h后,用Hank盐平衡液清洗顶室、底室,向顶室内加入100 μL异硫氰酸荧光素标记的葡聚糖(FITC-dextran,1 g·L-1),下室内加入500 μL Hank盐平衡液,37 ℃避光孵育2 h,期间每30 min从基底侧收集1次上清液,置于96孔板上,荧光分光光度计测定荧光强度,激发光波长为480 nm,发射光波长为520 nm,制作标准曲线并计算FITC-dextran浓度。每组设3个复孔。
2.7 Western blot法检测细胞内通路蛋白表达水平按“2.4”中方法处理细胞并给药,继续作用12 h后,胰酶消化并裂解细胞,离心收集上清液。BCA法测定蛋白浓度,各组均取30 μg蛋白,加等体积Buffer混合、上样,行SDS-PAGE电泳。200 mA电转2.5 h,PVDF膜室温封闭1 h。洗膜后移入分别含PERK、elF2α、ATF4和CHOP一抗(稀释比例为1 ∶1 000)的孵育盒中过夜;洗膜3次,转移到含二抗(稀释比例为1 ∶5 000)的孵育盒中孵育1 h;再次洗膜3次后用增强化学发光法检测阳性信号,分析各电泳条带的性质。

3 结果
3.1 GXD对应激态Caco-2细胞存活率的影响Tm干预Caco-2细胞12 h后,通过CCK-8法测定结果显示:MC组细胞的存活率为46.88%±6.09%,明显低于NC组的97.16%±4.21%,差异有统计学意义(P<0.01);GXD-L、GXD-M和GXD-H组的存活率分别为49.01%±3.86%、73.50%±8.94%和82.23%±7.70%,与MC组相比,GXD-M、GXD-H组存活率升高,差异有统计学意义(P<0.01)。见Fig 1。
3.2 GXD对应激态Caco-2细胞凋亡率和细胞周期分布的影响通过流式细胞术测定结果显示:① MC组细胞的凋亡率为29.95%±2.16%,明显高于NC组的3.13%±0.28%,差异有统计学意义(P<0.01);GXD-L、GXD-M和GXD-H组的凋亡率分别为19.02%±1.90%、16.33%±2.11%和10.26%±2.07%,与MC组相比,GXD-L、GXD-M和GXD-H组的凋亡率均显著降低,差异有统计学意义(P<0.05)。结果见Fig 1、2。② MC组G2期细胞的比例为5.24%±0.58%,明显低于NC组的31.12%±4.41%,差异有统计学意义(P<0.01),提示Tm诱导细胞处于内质网应激状态时,细胞被大量阻滞于有丝分裂早期,从而增加了细胞DNA及其修复系统暴露于外源性有害刺激的时间和几率,细胞易发生损伤和凋亡;GXD-L、GXD-M和GXD-H组G2期细胞的比例分别为13.90%±1.81%、18.63%±2.50%和24.97%±1.90%,与MC组相比,GXD-L、GXD-M和GXD-H组的G2期细胞比例均升高,差异有统计学意义(P<0.01),提示GXD的干预可缓解细胞周期阻滞,有利于细胞对损伤的DNA进行及时有效的修复,避免细胞的损伤和凋亡。见Fig 3。
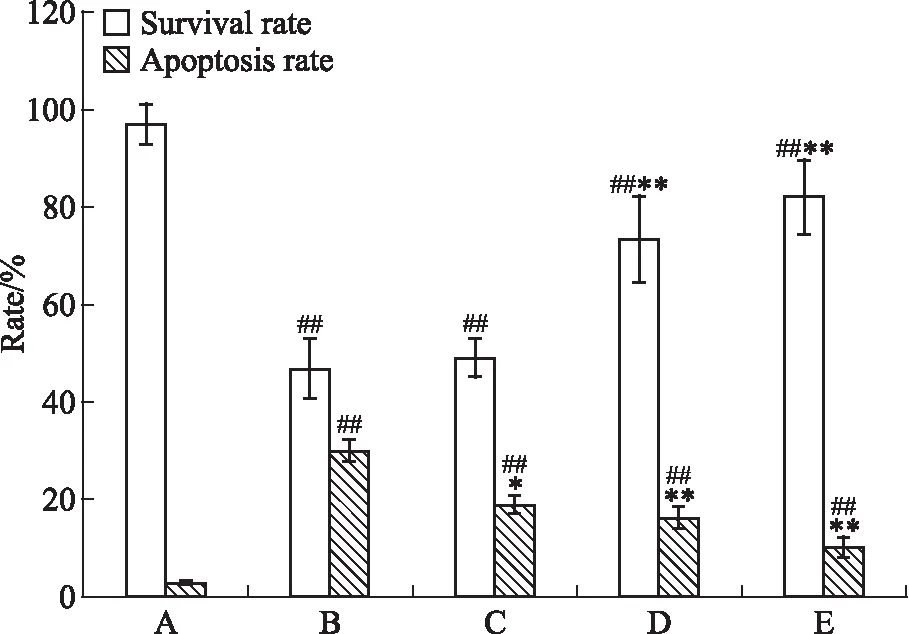
Fig 1 Comparison of survival rate and apoptotic rate of stress Caco-2 cells after intervention of different concentrations of GXDA:Normal control group;B:Model group;C:Low-dose GXD group;D:Medium-dose GXD group;E:High-dose GXD group.##P<0.01 vs normal control group;**P<0.01 vs model group.
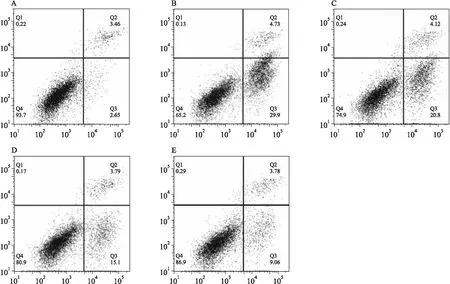
Fig 2 Effects of different concentrations of GXD on apoptosis of stress Caco-2 cells A:Normal control group;B:Model group;C:Low-dose GXD group;D:Medium-dose GXD group;E:High-dose GXD group.

Fig 3 Effects of different concentrations of GXD on cell cycle distribution of stress Caco-2 cellsA:Normal control group;B:Model control group;C:Low-dose GXD group;D:Medium-dose GXD group;E:High-dose GXD group.#P<0.05,##P<0.01 vs normal control group;**P<0.01 vs model group.
3.3 GXD对应激态Caco-2细胞屏障通透性的影响通过TEER检测和FITC-dextran法的结果显示:MC组的TEER值明显低于NC组,前者FITC-dextran的浓度明显高于后者,差异有统计学意义(P<0.01);与MC组相比,GXD-L、GXD-M和GXD-H组的TEER值不同程度地升高(P<0.01)、FITC-dextran的浓度不同程度地降低(P<0.05)。见Tab 1。
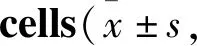
Tab 1 Effects of different concentrations of GXD on cell barrier permeability of stress Caco-2 n=3)
3.4 GXD对应激态Caco-2细胞内PERK-elF2α-CHOP信号通路关键蛋白表达水平的影响通过Western blot法的结果显示:MC组细胞内磷酸化的PERK(p-PERK)、磷酸化的elF2α(p-elF2α)、ATF4和凋亡关键蛋白CHOP的表达水平均明显高于正常对照组,差异有统计学意义(P<0.01);与MC组相比,GXD-M、GXD-H组中p-PERK、p-elF2α、ATF4和CHOP的表达水平均降低,差异有统计学意义(P<0.01)。见Fig 4。
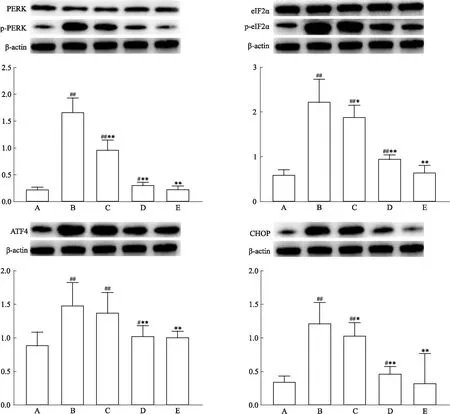
Fig 4 Effects of different concentrations of GXD on expression of PERK-eIF2α-CHOP signaling pathway in stress Caco-2 cellsA:Normal control group;B:Model group;C:Low-dose GXD group;D:Medium-dose GXD group;E:High-dose GXD group.#P<0.05,##P<0.01 vs normal control group;*P<0.05,**P<0.01 vs model group.
4 结论
既往普遍认为,UC的发生发展可能是包括遗传、环境、肠道菌群、免疫等在内的多因素共同作用的结果,其中免疫功能紊乱是下游共同的病理环节[2]。研究表明,肠黏膜屏障稳态破坏是引起UC发病和病情进展的重要原因。作为肠黏膜屏障主要结构和功能基础的IECs若发生过度凋亡,将导致屏障结构损伤、肠上皮通透性增高,多种外源性抗原物质异常暴露于固有层黏膜相关淋巴组织,刺激后者大量释放致炎因子,进而通过因子间网络化通讯使免疫反应级联放大,最终引起肠道炎症失控和结肠黏膜损伤[3-4]。在UC患者、UC动物模型中[11-13]以及体外ERS状态肠细胞模型[14]中均存在IECs大量凋亡和屏障通透性增高现象,提示IECs异常凋亡是引起肠黏膜屏障破坏、进而导致UC发生发展的重要环节。
ERS是指细胞在各种应激原的刺激下,其内质网合成加工蛋白质的生理功能障碍,大量未折叠或错误折叠蛋白在内质网腔中蓄积所启动的一系列细胞内稳态失衡过程[15]。静息状态下,ERS标志蛋白——葡萄糖调节蛋白78(glucose regulated protein78,GRP78)与内质网膜上的多种跨膜感受蛋白相结合,使后者处于失活状态。适度的ERS可使跨膜感受蛋白与GRP78发生解离而活化,进而激活“未折叠蛋白反应”这一保护性机制,有利于改善蛋白质的合成加工,避免应激细胞损伤死亡,体现了机体的自我稳定能力。但若应激过强或过久,未折叠蛋白反应不足以完全代偿缓解细胞损伤时,为维持细胞内稳态,活化的跨膜感受蛋白将进一步激活细胞凋亡程序,诱导应激细胞的程序性死亡[16]。目前已明确的ERS跨膜感受蛋白包括:PERK、活化转录因子6(the activating transcription factor 6,ATF6)和肌醇酶1(inositol requiring enzyme l,IREI),分别启动3条ERS特有的凋亡信号通路。其中PERK所启动的PERK-eIF2α-CHOP通路是十分重要的凋亡信号转导途径。与GRP78解离后的PERK通过同源二聚化而磷酸化激活自身,随后依次磷酸化elF2α、诱导转录活化因子4(the activating transcription factor 4,ATF4)的翻译表达,最终激活ERS独特的凋亡标志蛋白——CHOP的基因转录,后者通过调控死亡受体途径和线粒体途径两大经典方式激活凋亡效应子Caspase-3,执行细胞凋亡发生[17]。研究表明,PERK-eIF2α-CHOP信号通路参与甚至主导了如神经退行性病变、心肌病、2型糖尿病、慢性肾病、缺血/再灌注及部分化学中毒损伤等的许多疾病的病理生理过程[18];而抑制该通路上下游各关键蛋白的表达和/或活化水平,能够对抗凋亡,实现对ERS诱发损伤的保护,有效阻止上述疾病的发生发展。
在中医基础理论和实践中,UC病程长、缠绵难愈,多属本虚标实之证:其基本病机为正虚邪恋、寒热错杂,其中又以脾虚为主要矛盾。脾气虚弱、湿蕴胃肠的特点始终贯穿于溃病性结肠炎的各个阶段及各种证型中。因此,UC治疗原则应为健脾和中、平调寒热。GXD起源于《伤寒论》,拓展于《金匮要略》,该方能有效减轻UC的临床症状和肠镜下评分,提高生活质量,促进结肠黏膜的再生修复,改善肠黏膜屏障功能,降低复发率,与美沙拉嗪等药物共用能进一步提高临床疗效[8-10]。但GXD对肠黏膜屏障的保护作用是否通过调控PERK-eIF2α-CHOP信号通路、进而抑制IECs异常凋亡而实现,其中的具体机制和靶点亦不清楚,需要进一步探明。本课题组以Tm诱导法建立的体外IECs的应激态模型为研究对象开展实验,结果显示:① ERS激动剂Tm干预后,细胞的存活率明显下降、凋亡率明显上升、G2期细胞比例明显减少,单层细胞的TEER值明显降低,FITC-dextran浓度明显升高,提示ERS时细胞被大量阻滞于有丝分裂早期,并发生了严重损伤和大量凋亡,细胞屏障受损、通透性增加;而不同浓度的GXD作用后,各组细胞的存活率均上升、凋亡率均下降、G2期细胞比例均增加,单层细胞的TEER值均升高,FITC-dextran浓度均下降,提示GXD能缓解应激态细胞的周期阻滞,利于细胞及时修复损伤,减轻凋亡发生,保护细胞屏障和稳定屏障通透性。② ERS激动剂Tm干预后,细胞内p-PERK、p-elF2α、ATF4和ERS凋亡标志蛋白CHOP的表达水平均明显升高,提示ERS时PERK-elF2α-CHOP信号通路被激活而介导了凋亡信号的转导;而中、高剂量的GXD作用后,细胞内p-PERK、p-elF2α、ATF4和CHOP的表达水平均明显下降,提示GXD可通过抑制该通路的信号转导,从而发挥抗凋亡效应,保护IECs屏障。
上述研究结果阐释了GXD对UC的部分治疗机制:GXD可通过减少细胞的应激损伤和过度凋亡,从而保护IECs细胞屏障稳态,抑制PERK-eIF2α-CHOP信号通路的激活可能是其治疗靶点。GXD是否能同时影响ERS的其他特异性凋亡途径,其对IECs的其他损伤方式以及细胞旁途径是否有保护作用,仍需要后续更为深入的系列研究来明确。
猜你喜欢
江苏安全生产(2022年8期)2022-11-01
小资CHIC!ELEGANCE(2021年36期)2021-10-15
昆明医科大学学报(2021年8期)2021-08-13
艺术评鉴(2020年5期)2020-04-30
人大建设(2018年10期)2018-12-07
中国内镜杂志(2017年2期)2017-03-20
腹腔镜外科杂志(2016年10期)2016-06-01
中国病理生理杂志(2015年8期)2015-12-21
中国医疗美容(2015年1期)2015-07-12
医学研究杂志(2015年3期)2015-06-10
