
重氮苯与不同亲核试剂结合选择性:共价与非共价作用分析
2021-07-11 16:25王高博
高等学校化学学报 2021年7期
王高博,马 晶
(南京大学化学化工学院,介观化学教育部重点实验室,南京210023)
1 Introduction
The reaction between diazobenzene and various nucleophiles[Fig.1(A)],one of the important organic synthesis methods,has a history of more than a century.For example,this kind of reaction was applied in the preparation of fluorine-containing aromatic compounds,which occupies a large proportion in medicines and agrochemicals.However,the mechanism of such an important reaction,especially the binding selectivity of nucleophiles,has not been fully elucidated.
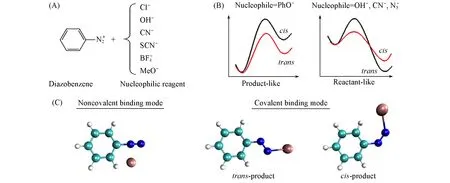
Fig.1 Schematic illustration of the reaction between diazobenzene and various nucleophiles(A),the proposal of cis and trans preference(B)and different binding modes(C)
From the middle of the last century to the 1980s,American chemist Swainet al.[1]and German chemist Zollinger[2,3]have made important contributions to the study of the mechanism of this reaction.An effective method of introducing F atoms into aromatic rings is the Balz-Schiemann reaction,in which N2is proposed to first leave as a leaving group.In the 1970s,Swainet al.[1]found that the first-order rate constant for the dediazoniation reaction changed less than 2%when the solvent changes from 80%to 105%H2SO4,where the concentration of nucleophiles drops rapidly.The 105%H2SO4means that 5%mole fraction sulfur trioxide is dissolved in pure sulfuric acid[2].That means nucleophiles is not involved in the formation of the transition state,and the rate-determining step is simply a dissociation process.However,the conclusion drawn from tetrafluoroborate diazobenzenes by Swainet al.[1]might not be general for a broad scope of reactions.
Zollinger[2,3]found that the binding sites of different nucleophiles with diazobenzene were different from each other.This phenomenon was ascribed mainly to kinetic factors.Zollinger proposed that when a transition state was“reactant-like”(e.g.,with OH-,),it tended to form acis-azo-isomer,and when a transition state was“product-like”(e.g.,with PhO-),atrans-isomer was preferred,as shown in Fig.1(B).Such a speculation is explanatory rather than predictive for reactions involving unknown reagents.Note that,as will be mentioned hereafter,the“binding”is just the process of combining diazobenzene and nucleophile,rather than a nucleophilic substitution reaction.
In this work,we use density functional theory(DFT)to study the electronic structures of complexes of six nucleophilic reagents(Cl-,OH-,CN-,SCN-,BF4-and MeO-)anchoring at three different binding sites in aqueous solution[Fig.1(C)].The conceptual density functional theory(CDFT)[5—7]is used to locate the possible binding sites.The CDFT has also been used in the quantitative description of nucleophilicity,electrophilicity and acid-base strength[8].Based on some qualitative analysis methods,such as average local ionization energy(ALIE),we proposed a descriptor,calledSTC,to predict the activation energy of the reactions.The insight gained in this work will be helpful to find better conditions for the important reaction in organic synthesis.
2 Computational Details
All calculations were carried out with the Gaussian 16 software package[9].Geometry optimization and frequency analysis were carried outviaDFT calculations with theoretical level of M06-2X[10]/Def2-TZVP[11].The implicit solvation model SMD[12]was used to model the solvent effect of aqueous solution.In addition,the Hirshfeld population analysis[13],electrostatics surface potential(ESP),ALIE analysis[14]and reduced density gradient(RDG)analysis[15]were performed with Multiwfn software[16]to establish the correlation between the electronic structure properties and the reactivity and selectivity of reactions.The visual molecular dynamics(VMD)[17]code was used for visualization of electron distributions of reactants.
3 Results and Discussion
The reaction process has two steps.The reaction is initiated from the approaching of nucleophile to diazobenzene substrate,in which step the covalent bond is not formed yet.As shown from ESP of diazobenzene[Fig.2(A)],the non-covalent interaction between two reactants is mainly dominated by electrostatic interaction.We did not plot the ESP surface of the nucleophiles because the volume of the nucleophiles is relatively small,and the negatively charged atoms are very clear.In the subsequent step,the chemical bond formation is occurred.We will present the detailed analysis of electronic structures of reactive species to understand the role of non-covalent and covalent interactions played in the modulation of reactivity.

Fig.2 ESP fitting surface(BWR colored,midpoint=0.77)(A)and atomic serial numbers(B)of diazobenzene
Since diazobenzene is a cation,the ESP is positive and difficult to be illustrated with normal color scheme,which will give a picture in dark blue.Thus,the ESP shown in Fig.2 is BWR colored with the selected Midpoint of 0.77,rather than 0.It can be seen from Fig.2 that the specific electrostatic region is lying around the diazo group.DFT calculations were widely used to evaluate qualitative chemical concepts such as chemical softness,S(which is the reciprocal of hardness,η).For the studied diazobenzene,an N-electron system,softness,S,is denoted as the reciprocal of a difference between ionization potential[obtained from the(N-1)-electron system]and electron affinity[from(N+1)-electron system].The local chemical softness of each atom of diazobenzene was also calculated,as shown in Table 1.The reduced Fukui function was then calculated with Hirshfeld charges of N-,(N-1)-,and(N+1)-electron systems.
It can be seen from Table 1 that the terminal N atom(called N13 in Fig.2)is the most favorable site for nucleophilic attack,followed by its neighboring N atom(i.e.,N12).The C2 linked to diazo group was not a favorable site for attack,which also implied that the pathway of forming a transition state through nucleophilic attack on C2 is unlikely to take place.
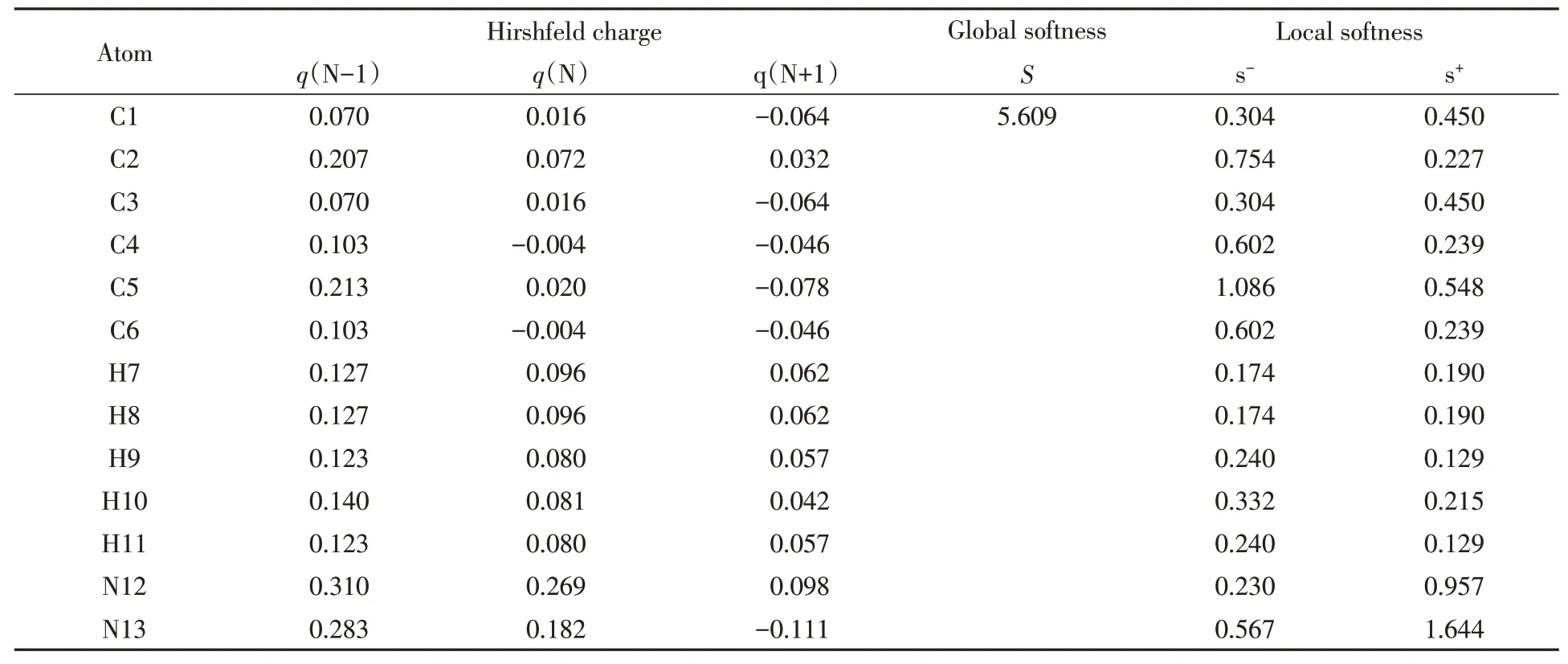
Table 1 Hirshfeld charges(q),global softness(S),and local softness for electrophilic(s-)and nucleophilic(s+)reactions for diazobenzene
It is recognized that reagents with high chemical hardness(or in other word,low softness)is not prone to occur the charge transfer reaction,but tends to form ionic bond with reagents with also high chemical hardness.To the other end,reagents with small chemical hardness tend to form covalent bonds with the reagents with also small chemical hardness.As shown in Fig.3,“hard”nucleophiles,such as BF4-(η=0.420)and Cl-(η=0.635)are more likely to be bound in the“Max-potential ring”of diazobenzene through electrostatic interactions;while“soft”nucleophiles as OH-(η=0.232),CN-(η=0.303)and MeO-(η=0.222)are prone to form covalent bonds with N13 atom to form azo structures intrans-orcis-configuration.The configurationsshown in Fig.3(A)and(B)are global minima.Some local minima were also located.For example,the configuration withbinding at edge side is 7942 J/mol higher than that on the top site in Fig.3(A).To further gain more information of the nature of non-covalent interaction,we took DFT-D3 dispersion correction into account.Forsitting in top side,the correction energy is only about 1672 J/mol.

Fig.3 Binding modes of diazobenzene with five different nucleophiles
It should be pointed out that tetrafluoroborate cannot attack C2 directly,but hover over the“Max-potential ring”.Then we want to know how the presence of tetrafluoroborate affects the dissociation process of diazobenzene.We carried out the reduced density gradient,reduced density gradient(RDG),analysis for tetrafluoroborate diazobenzene and get the scatter and isosurface diagram,shown in Fig.4.
As shown in Fig.4,one of the three fluorine atoms,which is in proximity to the benzene ring plane,has a strong electrostatics attraction with C2 atom,and the other two fluorine atoms are attractive to N12 atom.During the dissociation of diazobenzene,the positive charge was gradually concentrated on C2 atom,so the interaction between C2 and fluorine atoms was gradually strengthened,which significantly reduce the dissociation energy.At the same time,after the coplanarity of nitrogen molecule and benzene ring was destroyed,the N12—N13 bond was polarized by fluorine atoms,and was pulled to the direction of tetrafluoroborate(Fig.5).

Fig.4 RDG scatter and isosurface plot of diazobenzene tetrafluoroborate
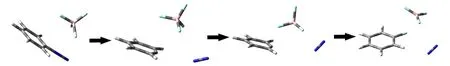
Fig.5 Decomposition process of diazobenzene with the help of tetrafluoroborate
On the other hand,for covalent binding,when nucleophilic reagents form covalent chemical bonds with N13 atom,there are two possible configurations,cis-ortrans-isomers.Zollinger has proposed that nucleophilic reagents experiencing the mechanism of“reactant-like transition state”tend to generatecis-isomers,while“product-like transition state”prefer to generate trans-isomers.In order to test the applicability of this hypothesis and specifically investigate the factors that determine the mechanism,we first calculated the intrinsic reaction coordinate curves of the formation ofcis-andtrans-isomers with cyanide ions and thiocyanate with sulfur and nitrogen atoms as attacking atoms,respectively.It was found that the attack of sulfur atom is difficult to formcis-isomers,which is different from the other five sets of nucleophilic reagents(Fig.6).

Fig.6 Intrinsic reaction coordinate plots of the reactions for CN-(A)and SCN-attacking(called ATK in short,B)diazobenzene
The relative energy ofcis-product is higher than that oftrans-one,so thecis-product is thermodynamically less stable than thetrans-product.However,the formation of transition state along thecis-path is much easier with lower activation energy,in agreement with Zollinger’s hypothesis.There is one exception.In the case of attacking of thiocyanate with N atom,the resultant transition state is much close to the product-like transition state,but the activation energy for reaching thecis-transition state is still lower than that for thetrans-transition state.This is probably because that the volume of sulfur atoms is too large,so the steric hindrance is large.However,it also can be seen that sulfur atom is obviously more favorable as attacking atom than nitrogen atom for a trans-path,which cannot be explained by the factor of steric hindrance.A detailed analysis is given as follows.
When acis-transition state is formed,due to the steric effect,the coplanarity of diazo structure and benzene ring will be destroyed.For example,when thiocyanate attacks with nitrogen atoms,the dihedral angle between diazo plane and benzene ring plane is 47.4°in transition state,and the conjugation is weakened to a considerable extent(Fig.7).From this perspective,the formation ofcis-transition state is thermodynamically unfavorable.However,there are many examples that nucleophilic reagents are indeed more inclined to form acis-transition state with diazobenzene.This means that some other energetically favorable factors could offset the energy increase caused by conjugation destruction.
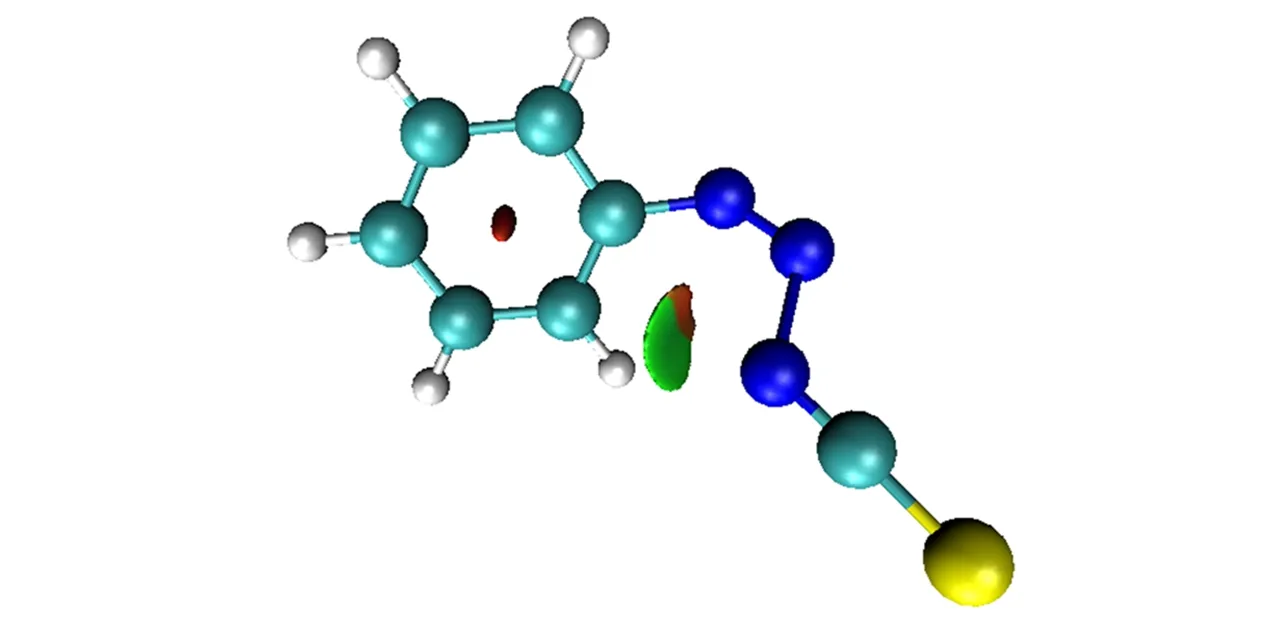
Fig.7 RDG isosurface of the transition state attacked by thiocyanate with nitrogen atom
We note that the nucleophilic reagent prefers to the formation ofcis-transition state which often has an attacking atom with high chemical hardness.Accor-ding to hard and soft acid and base(HSAB)theory,the energy lowering may originate from non-covalent interactions,that is,the ortho-hydrogen of the benzene ring may form a hydrogen bond with the attacking atom.We analyze the transition state of thiocyanate attacked by nitrogen atoms with RDG method.As shown in Fig.7,there is indeed an obvious attractive interaction(displayed in green color)between nitrogen atoms and the ortho-hydrogen atom,leading to the formation ofcis-product.
Finally,it is meaningful to correlate the activation energy barrier of nucleophilic attack with a descriptor.Taking the molecular volume or diameter to describe the steric hindrance effect,we calculated the minimum distance,lmin,and maximum distance,lmax,between the nucleophilic reagent binding sites and van der Waals surface.Then based on average local ionization energy,ALIE,we proposed a descriptor of trans-path,ST,as follows:

Forcis-path,we define theSCdescriptor:

The above two descriptors,STandSC,are combined into one parameter.The descriptor,STC,is defined asSTC=min{ST,SC}.In other words,ifST>SC,acis-path would be preferred,thenSTC=SC.STCis a good descriptor that can be used to predict the energy barrier of the reactions between nucleophilic reagents and diazobenzene(Fig.8 and Table S1,see the Electronic Supplementary Material of this paper).A good correlation between the activation energy barrier,ΔE,andSTCdescriptor is obtained.

Note that in fact in Fig.8,STandSC(or cis and trans path)are clustered into two different data groups.The detailed information of nucleophiles and reaction paths are shown in Table S1.
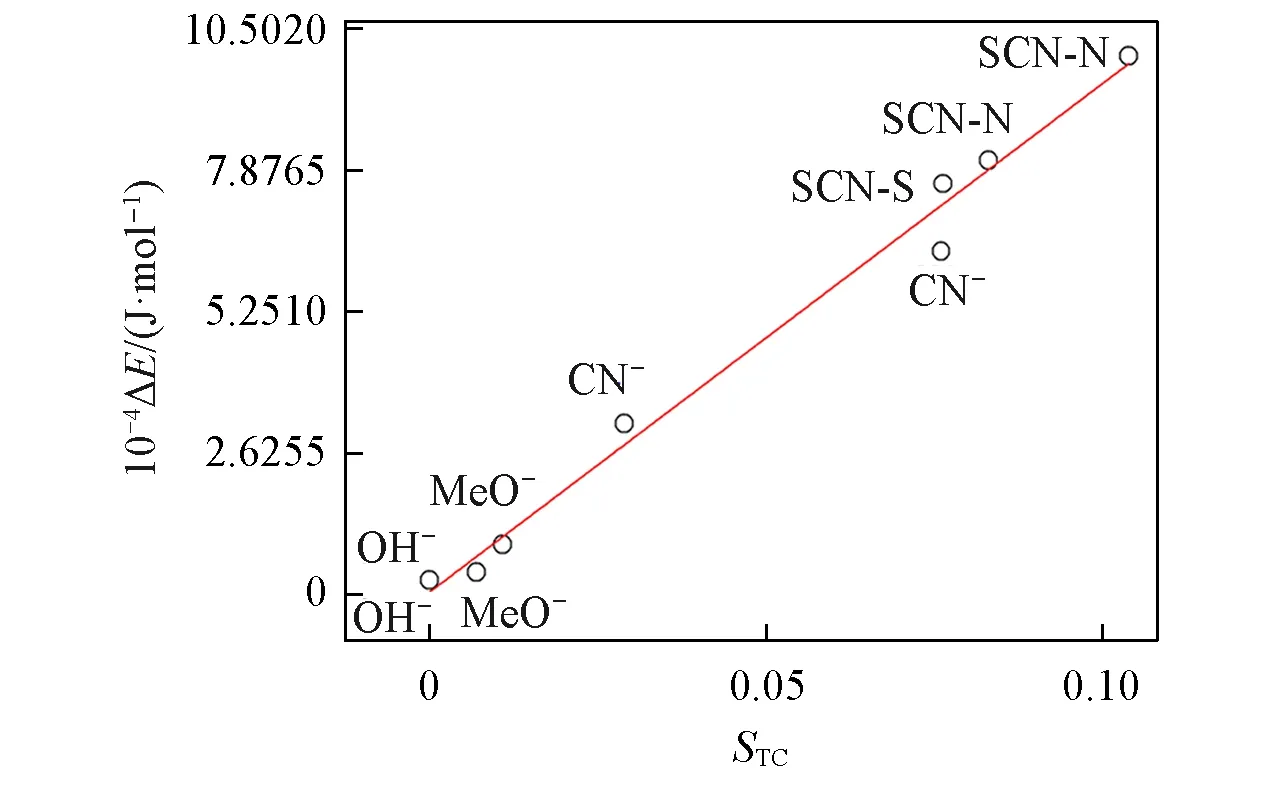
Fig.8 Relationship between the activation energy barrier,ΔE,and S TC(Pearson’s r=0.99413)
4 Conclusions
In this work,we predicted the binding positions of different nucleophiles on diazobenzene with ESP fitting and local chemical hardness calculation.The role of tetrafluoroborate in the dissociation of diazobenzene is revealed.We also found that the‘reactant/product-like transition state’hypothesis cannot always give correct predictions on the spatial selectivity,and thecis-path was mainly dominated by noncovalent interaction.Consequently,we proposed a descriptor,STC,to predict the activation energy barriers.Our results provide useful information for elucidating the reaction mechanism involving aromatic diazonium salts.
The supporting information of this paper see http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20210237.
This paper is supported by the National Natural Science Foundation of China(Nos.21873045,22033004).
猜你喜欢
云南化工(2021年9期)2021-12-21
化工职业技术教育(2021年5期)2021-11-09
潍坊学院学报(2021年2期)2021-07-22
化工职业技术教育(2021年1期)2021-03-15
化工职业技术教育(2020年4期)2020-01-19
食品科学(2019年14期)2019-07-26
东华大学学报(自然科学版)(2018年1期)2018-06-29
化工学报(2016年3期)2016-03-14
物理化学学报(2015年7期)2015-12-30
海南师范大学学报(自然科学版)(2015年2期)2015-12-23
