
基于模体中心结构的蛋白质复合物识别算法
2021-11-26 08:48夏雪飞王铭杰吴佳楠
吉林大学学报(理学版) 2021年6期
夏雪飞, 王铭杰, 吴佳楠
(1. 长春大学 量子信息实验室, 长春 130022; 2. 吉林交通职业技术学院 交通信息学院, 长春 130012)
蛋白质是生命活动的执行者, 生物体内蛋白质所有相互作用组成大规模网络, 即蛋白质相互作用网络PPI(protein-protein interaction network)[1], 可用含点集和边集的无向图表示. 蛋白质相互作用网络具有模块化特征, 模块内部的蛋白质有紧密的拓扑和功能联系, 这些模块被称为蛋白质复合物PC(protein complex)[2]. 在PPI网络中检测蛋白质复合物有助于探索生命活动过程中的未知蛋白质功能、解释特定的生物进程, 对于疾病诊断和药品研发具有重要意义[3].
通过实验获得的蛋白质复合物十分有限, 高通量技术产生了大量的蛋白质相互作用数据, 为使用计算方法识别蛋白质复合物提供了极大帮助. 通常方法是将蛋白质相互作用数据转化为用图描述的蛋白质网络, 蛋白质复合物对应图中的稠密子图, 采用图分析方法对蛋白质复合物进行识别, 例如: King等[4]提出了一种代价函数的聚类算法RNSC(restricted neighborhood search clustering), 该算法是一种典型的基于图划分方法; Palla等[5]提出了一种团渗透算法CPM(clique percolation method), 该算法是一种基于密度的局部搜索方法; 文献[6]中的G-N算法是一种基于层次聚类的方法. 近年, 人们基于核心-附属蛋白质的结构[7]提出了一些新型算法, 如COACH(core-attachment)[8]和Core方法(a novel core-attachment based method)[9]等. 由于在静态网络上挖掘蛋白质复合物无法充分体现功能模块的内在结构和动态属性, 因此一些研究人员结合动态生物信息构造蛋白质网络, 如Tang等[10]构建了动态蛋白质网络; Ouyang等[11]提出了一种时序平缓重叠复合物检测机制, 可在动态网络中检测瞬时的蛋白质复合物. 但目前多数方法都在复合物中心节点的选择上主要依赖于图密度或节点关联度等信息, 复合物的中心通常设定为一个单节点, 即使算法本身具备高效的子网挖掘性能, 而识别的蛋白质复合物却常不具备显著的生物学意义.
网络模体(network motif)是复杂网络演化的重要拓扑结构[12], 其代表了复杂系统中的重要功能单元, 定义为复杂网络中在不同位置重复出现的特定相互连接模式, 出现频次显著地高于随机期望[13]. 如果一个子图在实际网络中出现的频次高于随机网络中出现的平均频次, 则该子图为模体. 蛋白质相互作用网络中存在具有层次模块化特性的模体结构[14-15]. Wuchty等[16]研究表明, 模体成员蛋白比非模体成员蛋白在进化上更具有保守性, 研究结果暗示模体可能是网络中进化保守的拓扑单元; Lee等[17]研究表明, 不同的模体模式所承受的进化约束显著不同, 这种综合了拓扑和功能的模体模式能更有效地表征PPI网络中进化保守的拓扑单元, 具有重要的生物学意义.
鉴于网络模体具有的进化保守特性, 本文提出一种新的以网络模体作为核心节点组的蛋白质复合物识别算法MCSS(motif central structure searching). 首先, 通过模拟数据实验验证该算法的可行性及计算准确性; 然后, 将本文算法应用于真实蛋白质相互作用网络数据进行测试, 并对识别的复合物进行富集分析, 发现了一些具有显著生物学意义的蛋白质复合物, 验证了算法的有效性; 最后, 为更直观地表达模体中心结构的蛋白质复合物结构体征, 将平面型二维网络拓扑进行三维立体转化.
1 模型及相关定义
1.1 中心模体
在蛋白质相互作用网络中, 蛋白质对应节点, 蛋白质之间的相互作用对应节点之间的连线. 网络模体是指出现频次较高的子图模式. 图1为PPI网络中所有可能的3阶模体拓扑和4阶模体拓扑. 由于蛋白质相互作用无方向性, 所以对于3节点模体, 共有2种不同的连接模式, 而对于4节点模体, 共有6种不同的连接模式.
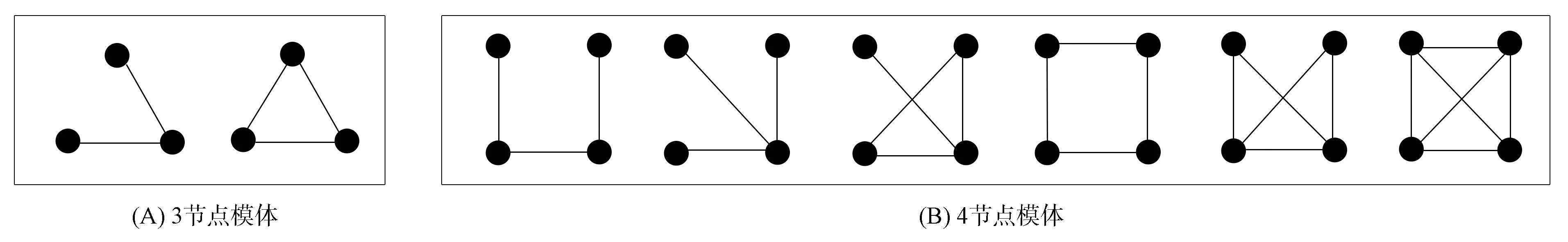
图1 PPI网络中的节点模体拓扑Fig.1 Node motif topology in PPI networks
1.2 中心模体三维转化
为更直观地表达本文算法所识别的复合物结构体特征, 本文尝试将二维的复合物网络拓扑进行三维转化, 转化方式如图2所示.

图2 蛋白质复合物立体转化示意图Fig.2 Schematic diagram of stereo transformation of protein complexes
1.3 相关定义
将整个蛋白质相互作用拓扑抽象为一个无向图, 即统计学中点与边的集合M=(P,E), 其中M为蛋白质相互作用网络图,P为M中的任意一个点,E表示M中任意两个点P的关联.
定义1(中心结构体饱和度) 具有相同节点的模体拓扑中, 节点间相互作用关系最多的模体结构饱和度最高, 如图1(B)中最后一个模体.
定义2(关联极限值) 限定计算范围为选定的中心节点Y和中心节点的直接邻居节点Xn共同构成中心结构体NL; 设n为一个以Y为中心节点的蛋白质复合物NL的蛋白质节点数,N为蛋白质复合物的相互作用极限关联值, 则饱和度最高的情形为
N=n(n-1)/2,
(1)
即复合物中每个蛋白质分子与其余蛋白质都有相互作用关系.
定义3(进化保守度) 为评估所识别蛋白质复合物的有效性, 本文为每个蛋白质复合物定义一个R值, 命名为进化保守度.R值越大则该蛋白质复合物被识别的概率越高. 设m为中心结构体NL中除中心节点外所有节点的实际相互作用关联数, 则R可表示为
R=(m/N)×100%.
(2)
2 蛋白质复合物识别算法
MCSS算法将模体作为蛋白质复合物的中心结构体, 并基于中心结构体进行二层节点扩充. 算法步骤如下:
1) 数据预处理, 构建PPI网络拓扑;
2) 模体中心节点筛选;
3) 中心结构体识别;
4) 二层节点扩充;
5) 蛋白质复合物识别.
对于步骤4)中二层节点的扩充计算, 主要采用基于距离测定的蛋白质复合物识别算法(IPC-DM)的理念[18], 调整后融入本文算法. 该算法核心思想如下: 用|Vk|表示复合物内部蛋白质总量; |Evk|表示v与k作用边数目, 则蛋白质v与复合物k的相互作用概率为

(3)
其中v和k需满足
SP(v,u)≤l,u∈Vc,
(4)
INvk≥Tin,
(5)
式中SP(v,u)表示蛋白质v和u之间的最短距离,Vc表示复合物内的蛋白质节点集,l为用户设置的正整数;Tin取值区间为(0,1).
算法包括3个子过程: 计算节点权重、选择种子和扩充簇. 先计算图中每条边的权重, 对每个节点所有连接边的权重求和, 得出节点权重, 并根据权重排序, 形成种子队列; 逐个扩充每个种子节点, 扩充的优先权由连接边数和边的权重和决定, 边数越多, 权重和越大, 优先权越高[18]. MCSS算法过程描述如下.
算法1MCSS算法.
输入:M=(P,E) //蛋白质相互作用网络
输出: var center_noden//中心节点n
var struct_listn//中心结构体除中心节点外的所有节点集合
var extension_listn//中心结构体扩展节点集合
varRn//中心节点n对应的邻居节点的实际关联值与关联极限值的比率
Result={ center_noden: {
struct_listn: [node1,node2,…,noden,
extension_listn: [node1,node2,…,noden],
Rn}}
Program:
1) func neibhbor(node) //计算当前节点node的邻居节点集合
2) varRmax//中心节点的邻居节点的关联极限值
3) var node //普通节点
4) var neib_node //中心节点的邻居节点集合
5) var center_node //中心节点
6) for center_nodeiinMdo
7)n=length(neib_node) //中心节点的邻居节点数
8)m=length(neib_edage) //中心节点的邻居节点的关联数
9) ifn< 3 do
10) continue
11) else
12)Rmax=(n2-n)/2 //中心节点的邻居节点的关联极限值
13) Result[center_nodei].Ri=m/Rmax//关联比率
14) Result[center_nodei].struct_listi=neib_node //中心节点的邻居节点集合
15) endif
16) for nodejin Result[center_nodei].struct_listi
17) append(Result[center_nodei].extension_listi, neibhbor(nodej))
18) end for
19) end for
20) return Result.
3 实 验
3.1 模拟数据实验
创建模拟数据集SimuPPIData1用MCSS算法进行计算, 共得到32个有效复合物, 筛选保守度排名前10的复合物(饱和度大于50%)转化为三维结构, 如表1所示, 其中红色节点和黄色节点共同组成中心结构体, 蓝色节点为二层扩展节点,R值为进化保守度.
为验证MCSS算法的计算有效性, 参照PPI数据结构特征, 创建模拟数据集SimuPPIData2, 抽取部分数据生成PPI网络拓扑如图3所示. 图3中包含了8个特征显著的中心结构体, 4个高饱和度, 4个低饱和度, 经计算均被算法识别.

表1 SimuPPIData中排名前10的复合物结构
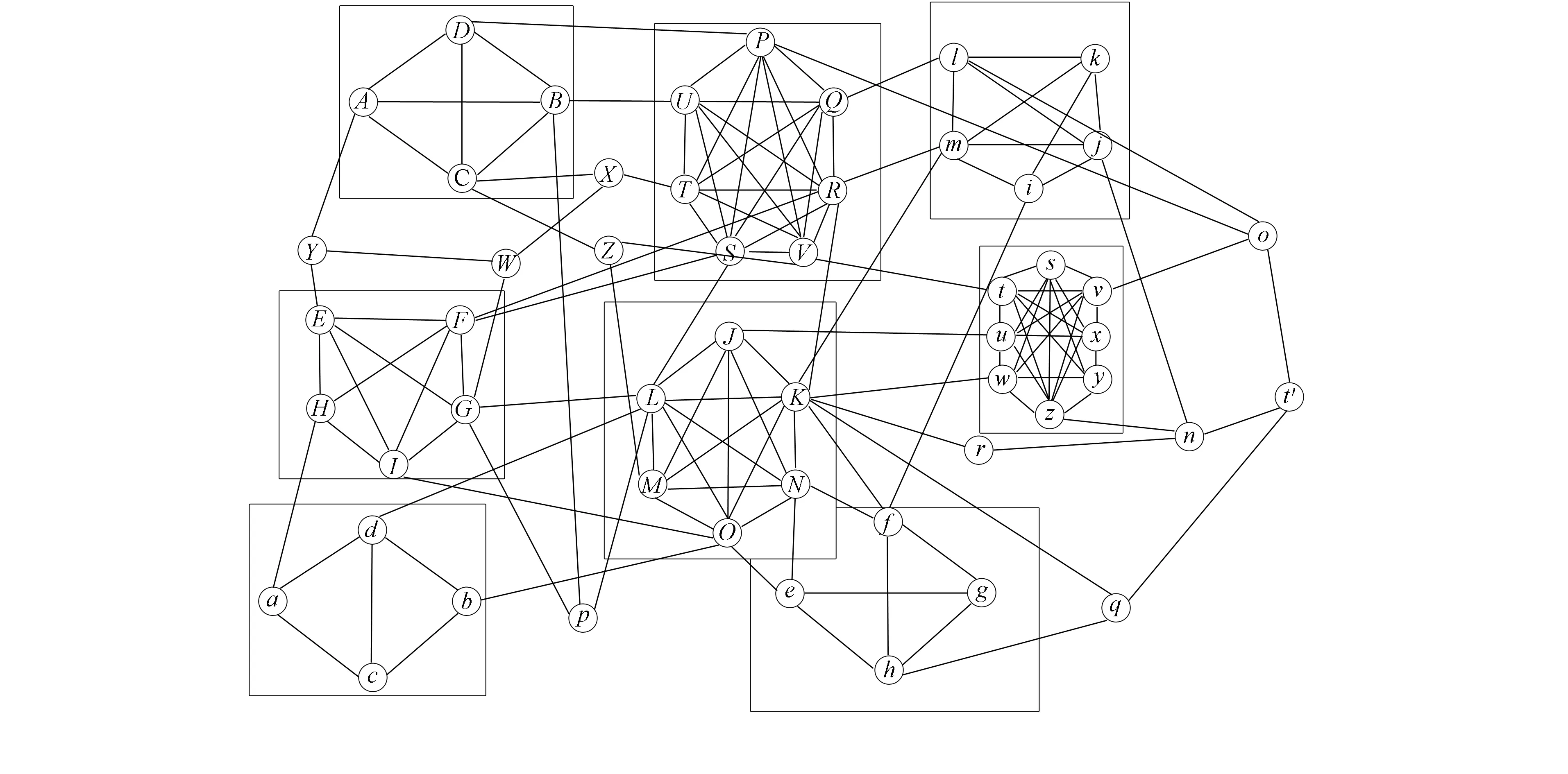
图3 SimuPPIData网络拓扑Fig.3 SimuPPIData network topology
3.2 真实数据实验
为验证算法的有效性, 本文使用酵母蛋白质数据对算法进行测试验证, 实验数据源于STRING数据库(https://string-db.org), 整组数据共9 687个节点, 168 974对关联, 经MCSS计算, 共识别出复合物集团1 128个, 对排名前5的复合物进行富集分析, 结果列于表2.

表2 排名前5的酵母复合物数据
生物数据库系统会为每个蛋白质复合物集团返回一个p值, 如果p值较小, 则表示该集团具有显著的生物学意义.p值计算公式为
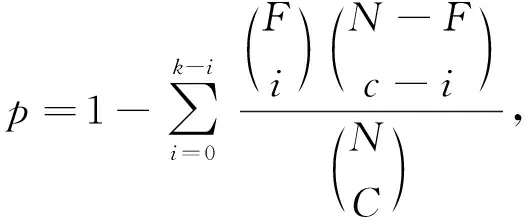
(6)
其中N为蛋白质相互作用网络的规模,C为蛋白质复合物中的蛋白质数,k为蛋白质复合物中含有某个功能的蛋白质数量,F为蛋白质相互作用网络中含有该功能的蛋白质数量[18]. 实验结果表明, 进化保守度R值越大集团p值越小, 对应的集团生物学意义越显著, 间接有效地证明了本文算法的有效性.
综上所述, 本文提出了一种新的以网络模体作为核心节点组的蛋白质复合物识别算法, 该算法根据蛋白质相互作用网络的拓扑特性, 将模体作为蛋白质复合物的中心结构体, 并基于中心结构体进行二层节点扩充, 能准确有效地识别蛋白质复合物. 通过模拟数据和酵母蛋白数据验证了算法的计算准确性和识别有效性.
猜你喜欢
橡塑技术与装备(2022年3期)2022-03-17
新世纪智能(数学备考)(2021年9期)2021-11-24
当代陕西(2019年15期)2019-09-02
智能计算机与应用(2019年1期)2019-01-11
中成药(2018年7期)2018-08-04
中成药(2018年3期)2018-05-07
学苑创造·A版(2018年11期)2018-02-01
中成药(2017年5期)2017-06-13
读者(2017年5期)2017-02-15
自动化学报(2016年5期)2016-04-16
