
组蛋白赖氨酸甲基转移酶抑制剂研究进展
2021-12-14 06:43李凌木唐海郭叶韩开林
药学进展 2021年9期
李凌木,唐海,郭叶,韩开林**
(1.内蒙古科技大学包头医学院药学院,内蒙古 包头014060;2.天士力控股集团有限公司研究院化学药品开发中心,天津300410)
1 组蛋白赖氨酸甲基转移酶
1.1 组蛋白赖氨酸甲基转移酶介绍
组蛋白甲基化转移酶(histone methyltransferase,HMT)的基因改变诱导了人类肿瘤疾病的发生,组蛋白赖氨酸甲基转移酶(enhancer of zeste homolog 2,EZH2)作为HMT重要成员,与胚胎外层发育蛋白(embryonic ectoderm development,EED)、ZESTE12同源物1抑制因子2蛋白(suppressor of zeste 12,SUZ12)、人AE结合蛋白2(AE binding protein 2,AEBP2)和视网膜母细胞瘤相关蛋白46/48(retinoblastoma associated protein 46/48,RbAp46/48)构成了起始复合物——多梳蛋白复合体2(PRC2)的催化亚单位[1-3],主要催化组蛋白H3上27位点赖氨酸(lysine 27 in histone H3,H3K27)甲基化,从而导致基因沉默[4],其在控制参与调节细胞增殖分化方面扮演重要的角色(见图1)。最新研究表明EZH2还与抑癌基因和上皮-间充质转化(epithelial-mesenchymal transition,EMT)相关基因如p16INK4a和上皮细胞钙黏蛋白基因的抑制有关[5]。据报道,EZH2的突变或过表达已在多种肿瘤中被发现,例如,在淋巴瘤中的功能获得性突变和过度表达。同时,在乳腺癌[6]、膀胱癌[7]、卵巢癌[8]、肝癌[9]、前列腺癌[10]、小细胞肺癌(SCLC)、黑色素瘤[11]、胶质母细胞瘤和儿童胶质瘤[12]等实体瘤中EZH2表达明显高于癌旁组织且与疾病进展和不良预后相关。相反在骨髓增生异常综合征(MDS)、MDS/骨髓增殖性疾病(MPN)、骨髓纤维化、急性髓细胞白血病(AML)和急性T-淋巴细胞白血病(T-ALL)中的EZH2功能丧失[13-16]。鉴于其在肿瘤发生和进展中的作用,EZH2已成为潜在的抗肿瘤治疗靶点[1,17]。

图1 起始复合物——多梳蛋白复合体2核心成分Figure 1 Core components of initiation complexpolycomb protein complex 2
1.2 组蛋白赖氨酸甲基转移酶致癌机制
在正常干细胞中,EZH2通过抑制谱系指定因子[18-20]来抑制分化,与非肿瘤细胞系相比,其在肿瘤干细胞(CSC)群体中表达水平更高,且必不可少[21]。EZH2和PRC2在发育过程中的功能可能不是促进生长或分化本身,而是抑制转录过程[22]。因此,抑制EZH2功能具有高度的细胞特异性。鉴于EZH2作为转录调节因子的作用,人们一直致力于寻找其驱动转化的下游目标。研究证明EZH2位于细胞周期调控的视网膜母细胞瘤——核因子E2(E2 factor,E2F)通路的下游,同时也参与视网膜母细胞瘤增殖基因表达和E2F驱动的增殖过程[23]。
除在组蛋白修饰和转录调控中的作用外,EZH2还可以甲基化非组蛋白底物。EZH2结合并甲基化信号转导及转录活化因子3(signal transducers and activators of transcription,STAT3),从而提高胶质母细胞瘤中CSC的致瘤性;在前列腺癌模型中,EZH2已经被证明甲基化雄激素受体(AR)并调控AR向EZH2结合的位点富集[24-25]。综上所述,EZH2除了具有H3K27组蛋白甲基转移酶和转录抑制因子的作用外,还能够甲基化非组蛋白底物如STAT3、锌指转录因子(GATA binding protein 4,GATA4)、踝蛋白(Talin)和维甲酸相关孤儿受体α(retinoic acid related-orphan receptors α,RoRα),从而促进转录沉默和转录激活;EZH2在转录激活中可以不依赖于PRC2,是AR相关复合体、核因子κB(nuclear factor kappa-B,NF-κB)、tcf/β-连 环 蛋白(tcf/β-catenin)、增殖细胞核抗原(proliferating cell nuclear antigen,PCNA)、β-连环蛋白(β-catenin)、雌激素受体α(estrogen receptor,ERα)等转录因子的共同激活因子[26]。
2 组蛋白赖氨酸甲基转移酶抑制剂
作为表观遗传领域热门靶点,EZH2抑制剂已经成为抗肿瘤药物开发的热点之一。其通过抑制EZH2过表达及突变,达到抑制肿瘤发生和发展的目的。
2.1 组蛋白赖氨酸甲基转移酶抑制剂分类
根据作用机制不同,EZH2抑制剂主要分为:1)抑制甲基转移酶的S-腺苷-l-高半胱氨酸水解酶(S-adenosyl-l-homocysteine hydrolase,SAH)水解的酶抑制剂;2)与PRC2复合物底物S-腺苷甲硫氨酸(S-adenosylmethionine,SAM)竞争性结合的竞争抑制剂;3)通过稳定EZH2 α-螺旋结构域破坏EZH2和EED蛋白相互作用的小核酸类药物,代表药物有ION674。
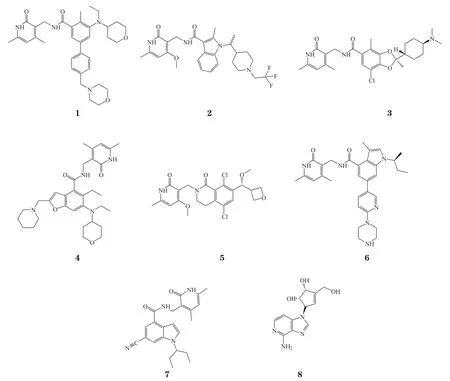
晶体学研究表明,在PRC2复合物的SAM竞争性抑制剂结构中,2-吡啶酮部分是酶抑制的关键,因为其占据了酶结合口袋中共底物SAM(或副产物SAH)的部分位置,解释了所有2-吡啶酮抑制剂表现出的SAM竞争作用机制。2-吡啶酮弹头连接到支撑子结构,按支撑子结构主要可分为2类:1)简单的单环(杂环)芳香环如tazematostat(1)等,2)双环杂芳环,如CPI-1205(2)、DS-3201(3)、SHR2554(4)、PF-06821497(5)、GSK-126(6)EL1(7)和DZNep(8)等。
2.2 组蛋白赖氨酸甲基转移酶抑制剂的临床研究
作为表观遗传领域新兴的抗肿瘤药物,目前已有1个药物已上市,及多个EZH2抑制剂处于临床研究阶段,表1总结了目前已上市及进入临床试验阶段的EZH2抑制剂。
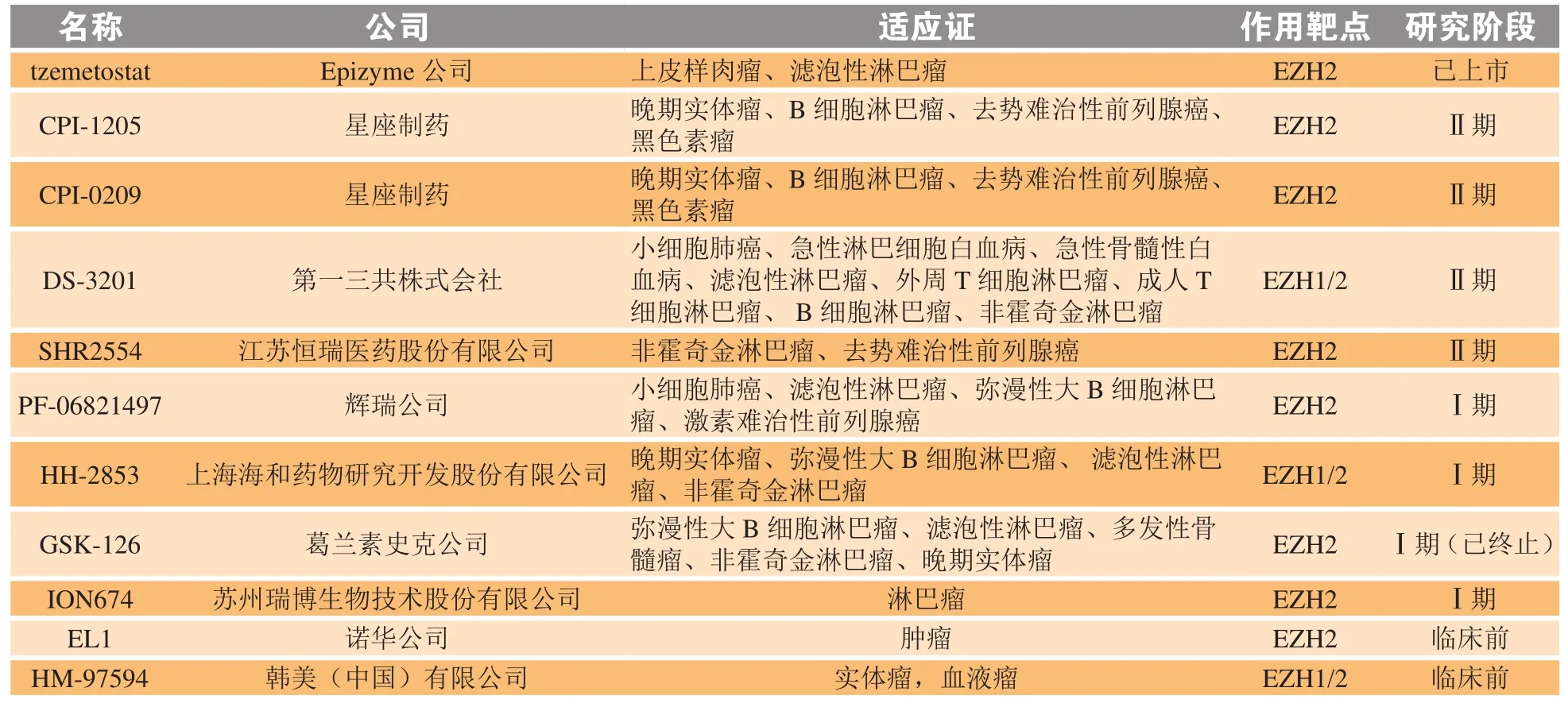
表1 组蛋白赖氨酸甲基转移酶抑制剂临床研究进展Table 1 Clinical research progress of EZH2 inhibitors
2.2.1 Tazemetostat Tazemetostat是Epizyme公司研发的一款Ⅰ类EZH2抑制剂,用于治疗滤泡性淋巴瘤和不适合完全切除的转移性/局部晚期上皮样肉瘤(epithelioid sarcoma,ES)患者[27],已于2020年1月在美国上市。
体外研究显示,tazemetostat抑制野生型EZH2抑制常数(Ki)为(2.5f0.5)nmol · L-1,对 Y646F 突变型淋巴瘤细胞株 WSU-DLCL2 的 IC50为 32 nmol · L-1[28]。体内研究显示,tzemetostat在体内能阻止肿瘤细胞的生长,并延长异种原位移植模型的存活时间。将3种不同基因的人肿瘤细胞(D556、D425和ONS-76)植入小鼠小脑,D556组、D425组口服2周、ONS-76 组 口 服 4 周 tzemetostat(350 mg · kg-1),Kaplan-Meier曲线显示,tzemetostat治疗显著提高了3种模型动物中位存活时间,D556组从22.5 d增加到29 d(p= 0.04),ONS-76组从48 d增加到58.5 d(p= 0.000 8),D425组从20.5 d增加到27 d(p= 0.006)[29]。
Ⅰ期临床研究中64例患者中的23例出现3级或更严重的不良事件(AE),6例出现3级或更严重的与治疗相关的AE。AE造成12例患者(19%)发生药物中断,1例患者在最高剂量1 600 mg时产生4级血小板减少的剂量限制性毒性。并确定了tzemetostat的Ⅱ期临床试验中800 mg(bid)口服的剂量,在这项研究中评估了tzemetostat的抗癌活性,该研究显示38%的B细胞非霍奇金淋巴瘤患者和5%的实体瘤患者均表现出持久的客观反应[30]。
Ⅱ期临床研究中tzemetostat在r/r滤泡性淋巴瘤(FL)或r/r弥漫性大B细胞淋巴瘤(DLBCL)患者中表现出高度的临床活性和持久的反应且具有良好的安全性。在EZH2突变的淋巴瘤和野生型EZH2中证实了tzemetostat的疗效:39例EZH2基因突变激活的r/r FL患者客观缓解率(ORR)为74%,完全应答率(CR)为10%。在53例可评估的EZH2野生型患者中,ORR为34%,CR为6%[31]。在r/r DLBCL患者中,突变型和野生型亚组的ORR均为17%,并且在突变型自回归模型(auto-regressive model,ARM)中可以看到更长的应答持续时间[32]。最近的一项更新数据显示43例EZH2突变患者的ORR为77%[33]。
2.2.2 CPI-1205 CPI-1205是星座制药公司研发的一款Ⅱ类EZH2抑制剂,目前处于Ⅱ期临床研究阶段,通过亲和闪烁法测定CPI-1205对野生型EZH2(WT)的 IC50为 2.2 nmol · L-1,对突变型 EZH2(641N MT)的 IC50为 3.1 nmol · L-1,海拉细胞系(HeLa)中 H3K27me3 的 IC50为 32 nmol · L-1[34]。给 荷瘤的雌性CB-17 SCID小鼠经口给药CPI-1205(160 mg ·kg-1,bid),连续25 d。2周内小鼠肿瘤消退,到第25 d结束时,记录到明显的肿瘤生长抑制且对重复给药的耐受性良好,没有明显的体质量减轻。最后1次给药1 h后的血浆和肿瘤药动学分析显示,CPI-1205在血浆和肿瘤组织中表现出较高浓度[34]。
目前,CPI-1205正进行2项临床试验,其中1项2017年11月15日启动的Ⅰb/Ⅱ期临床试验,旨在评估CPI-1205与苯扎鲁胺或阿比特龙/强的松联合治疗转移性去势抵抗性前列腺癌(mCRPC)患者。Ⅰb期研究中纳入了36例患者:20例服用CPI-1205+阿比特龙,16例服用CPI-1205+苯扎鲁胺。数据显示除2例患者无前列腺特异抗原(PSA)值外,剩余34例患者PSA值均下降50%以上,其中2例服用CPI-1205+阿比特龙和3例服用CPI-1205+恩扎鲁胺患者PSA值下降超过80%;最常见的与治疗相关的AE是疲倦、腹泻和恶心,CPI-1205与苯扎鲁胺联合使用时出现与治疗相关的3级不良反应,包括乏力、恶心和谷丙转氨酶(ALT)升高(1例出现3级不良反应;占总受试者的6.3%),与阿比特龙联合使用时出现与治疗相关的3级不良反应,包括乏力和ALT升高(1例,5%);确定了Ⅱ期临床试验中800 mg(tid)口服的剂量[35]。Ⅱ期临床试验旨在评估CPI-1205与大鼠肉瘤病毒同源癌基因激酶(rat sarcoma viraloncogenehomolog,RAS)抑制剂联合作为治疗mCRPC患者的二线药物的作用。另一项是2017年12月14日启动的Ⅰ/Ⅱ期试验,旨在评估CPI-1205 +伊匹单抗联合治疗晚期实体瘤患者(NCT03525795)[36]。
2.2.3 CPI-0209 CPI-0209是星座制药公司研发的第2代Ⅱ类EZH2抑制剂,旨在延长保留时间,与第1代抑制剂相比,其不会产生自身诱导作用而使半衰期缩短。目前处于Ⅱ期临床研究阶段。体外针对 EZH2(WT)的 IC50为 0.18 nmol · L-1;对 HeLa细胞中 H3K27me3 的 IC50为 2.3 nmol · L-1;细胞色素P450 3A4酶(CYP3A4)上调水平降低50%,体外数据较CPI-1205均提高90%以上。在淋巴瘤异种移植模型中,经口给药CPI-0209(25 mg · kg-1,qd)使肿瘤体积缩小,并且在治疗结束后表现出持久的客观反应。从第5 d至第19 d,肿瘤的体积约从250 mm3减少到0 mm3,而对照组的肿瘤体积从第5 d的250 mm3增加到第19 d的800 mm3和第29 d的1 750 mm3(治疗结束)。在雄激素受体剪接变异体7(androgen receptor splice variant 7,AR-V7)+前列腺癌异种移植模型中,口服 CPI-0209(100 mg · kg-1,bid)显示出高度活性[37]。2019年9月18日CPI-0209单药及联合治疗晚期实体瘤的Ⅰ/Ⅱ期研究正式启动(NCT04104776),将患者分为2组,一组单独使用CPI-0209,一组与伊立替康联合使用,截至2021年4月该项研究仍在推进中[38]。
2.2.4 Valemetostat Valemetostat(DS-3201)是第一三共制药公司研发的一款新型的Ⅱ类EZH1/2双重抑制剂,目前处于Ⅱ期临床研究阶段。它能同时抑制EZH1/2的酶活性,对多种非霍奇金淋巴瘤(NHL)细胞,如DLBCL、套细胞淋巴瘤和外周T细胞淋巴瘤(PTCL)均有抗增殖作用,细胞活性GI50小于100 nmol · L-1,此外,体外剂量递增研究表明,在SMARCB1(SNF5)突变的恶性横纹肌样瘤(G-401)、ARID1A突变的卵巢透明细胞癌(TOV-21G)和BAP1突变的间皮瘤(NCI-H226)的细胞模型中,DS-3201均显示出较高的抗增殖活性(IC50< 100 nmol · L-1)[39]。
Ⅰ期临床数据显示口服DS-3201b的ORR为53%(8/15),CR1例,部分缓解(PR)7例,200 mg(qd)和300 mg(qd)剂量时出现3例4级血小板计数下降,1例3级贫血和1例3级肺炎的严重AE。除血液毒性外,其他不良反应还包括排便障碍、腹泻、鼻咽炎、脱发、皮疹、食欲下降和皮肤干燥等[40]。2019年最新研究显示,有13例患者接受了T细胞扩增队列的治疗,其中7例成人T细胞白血病/淋巴瘤(ATLL)患者和6例PTCL患者都接受了200 mg的缬氨蝶呤治疗。在登记到Ⅰ期和扩展队列的ATLL患者中,ORR为44 %,9例患者中有5例继续治疗超过12周[41]。2019年11月一项名为DS-3201治疗复发或难治性ATLL的Ⅱ期临床研究在美国和日本被批准(NCT04102150);2020年8月DS-3201和伊匹单抗联合治疗转移性前列腺癌、尿道上皮细胞和肾细胞癌的Ⅰb期临床试验在美国被批准(NCT04388852),以上2项临床试验截至2021年4月均处于患者招募中。
2.2.5 SHR2554 SHR2554是江苏恒瑞医药股份有限公司开发的一款口服Ⅱ类EZH2抑制剂,适应证主要为非霍奇金淋巴瘤、去势难治性前列腺癌等,目前处于Ⅱ期临床研究阶段。体外研究显示,SHR2554对弥漫大B细胞淋巴瘤细胞株(Pfeffier)IC50为 2 nmol · L-1;对突变 EZH2(A677G)的 IC50为6.4 nmol · L-1。2018年8月于北大肿瘤医院启动了SHR2554治疗复发/难治性淋巴瘤的Ⅰ期临床试验(NCT03603951);2018年11月,复旦肿瘤医院启动SHR2554单独/联合SHR3680治疗mCRPC的Ⅰ/Ⅱ期临床试验(NCT03741712);2020年5月复旦肿瘤医院启动了治疗转移性乳腺癌的Ⅱ期临床试验(NCT04355858)。2020年9月,解放军总医院启动了SHR2554联合SHR1701治疗晚期实体瘤和B细胞淋巴瘤的Ⅰ/Ⅱ期临床试验(NCT04407741)。
2.2.6 PF-06821497 PF-06821497是辉瑞公司研发的一种有效的具有选择性的EZH2Ⅱ类抑制剂,目前处于Ⅰ期临床研究阶段。2018年4月17日该化合物在一项多中心的Ⅰ期研究(NCT03460977)中进行评估,研究对象为去势抵抗性前列腺癌(CRPC)患者(PF-06821497+苯扎鲁胺)、小细胞肺癌患者(PF-06821497+顺铂或卡铂+依托泊苷)和FL患者[42]。
体外研究发现PF-06821497对野生型和突变型Y641N-EZH2 的Ki均小于 0.1 nmol · L-1[43],并具有较好的EZH2抑制活性、亲脂配体效率(LIPE)、体外代谢稳定性和通透性。其体外多层线性模型(hierarchical linear model,HLM)稳定性为每毫克蛋白39μL · min-1,在体外人体肝细胞评估中稳定性为每百万细胞12μL · min-1,晶型的热力学溶解度为315 mg · L-1(在25℃的含0.5%羧甲基纤维素和0.1%聚山梨酯80的水中)。针对EZH2的野生型和Y641N突变体,miR-23a靶结合的半衰期分别为22和6 h。
体内研究发现,给携带Karpas-422 DLBCL肿瘤异种移植瘤(包含Y641N EZH2突变)的小鼠口服 PF-06821497(bid),口服剂量为 100 mg · kg-1可显著抑制肿瘤生长或口服剂量为300 mg · kg-1时肿瘤消退,每日皮下注射100 mg · kg-1剂量时,效果更好,且对体质量影响较小[43]。
2.2.7 HH-2853 HH-2853是上海海和药业研发的一款口服Ⅱ类EZH1/2双重抑制剂,用于晚期实体瘤,D弥漫性大B细胞淋巴瘤,滤泡性淋巴瘤等患者的治疗,目前处于Ⅰ期临床研究阶段。体外研究显示,HH-2853在野生型EZH2和突变型EZH2 (Y641F)中IC50值均小于100 nmol · L-1。2020年3月获美国FDA新药临床研究申请(investigational new drug,IND)默示许可,2020年8月其一项名为HH-2853安全性和临床药效的Ⅰ期临床试验(NCT04390737)在我国获批准,旨在评估EZH1/2抑制剂HH-2853在Ⅰ期及剂量递增中对复发/难治性非霍奇金淋巴瘤或晚期实体肿瘤患者的安全性、耐受性、药动学和临床疗效。确定最大耐受剂量(MTD)和推荐的Ⅱ期剂量(RP2D),截至2021年4月处于患者招募期[44]。
2.2.8 GSK-126 GSK-126是葛兰素史克公司研发的一款Ⅱ类EZH2抑制剂,在Ⅰ期临床试验结束后停止,体外研究发现GSK-126抑制皮肤淋巴瘤(B-NHL)中H3K27的三甲基化,而与EZH2激活突变的存在与否无关[45]。像B-NHL细胞系一样,GSK-126在AML和T细胞系中抑制H3K27me3的浓度比停止增殖和诱导凋亡所需的浓度低90%以上;4个EZH2GOFmuB-NHL细胞系中的3个(KARPAS-422、SU-DHL-6、WSU-DLCL2) 在 用GSK-126 处理后停止增殖,IC50为 2 ~ 10 nmol · L-1;低 浓 度 的 GSK-126(400 nmol · L-1) 不 仅 减 少 了DLBCL细胞系PFEIFFER中H3K27的甲基化,也导致了生长停滞[46]。体内研究发现,在负荷KARPAS-422和Pfeiffer异种移植的小鼠体内,GSK126 经 口 给 药 150 mg · kg-1· d-1时 降 低 总 体H3K27me3水平,增加基因表达,从而引起显著的肿瘤消退[47]。
Ⅰa期临床研究的数据显示,入组的41例患者(实体瘤21例,淋巴瘤20例)均发生至少1例AE。最常见的不良反应是乏力(22/41,53.7%)和恶心(20/41,48.8%)。12例患者(32%)经历了严重的AE。服用3 000 mg GSK-126的7例患者中有2例出现了肝转氨酶剂量限制性升高;因此2 400 mg被确定为MTD。使用剂量比例评估的药代动力学研究表明,GSK-126用量越大受试者出现限制性毒性的比例越大。然而,在第15 d,2 400 mg以下的所有剂量的谷值都低于体外蛋白质结合调节的抑制三甲基化的IC50(H3K27me3)。在耐受剂量下不能实现生物有效暴露,抗癌活性极低。说明GSK-126不是针对EZH2难治性/复发性实体和血液恶性肿瘤患者的可行药物[48]。
2.2.9 ION674 ION674是苏州瑞博生物技术股份有限公司从美国小核酸公司Ionis Pharmaceuticals,Inc引进的一款小核酸类EZH2抑制剂,用于淋巴瘤的治疗,目前处于临床前研究阶段。体外研究显示,此药对EZH2基因敲除HeLa细胞系、纯合子细胞(MZ-3776)的 IC50为 20 ~ 60 nmol · L-1、对人皮肤鳞癌细胞(A431)的IC50< 60 nmol · L-1、对人神经母细胞瘤细胞(SHSY)的 IC50< 500 nmol · L-1、对神经母细胞瘤细胞Kelly的IC50< 80 nmol · L-1。在CD1小鼠中测试化合物耐受性良好,肝功能、肾功能指标正常;组织质量较空白组无明显变化。2020年在美国获批进入Ⅰ期临床研究阶段。
2.2.10 EL1 EL1是诺华公司研发的一款Ⅱ类EZH2的SAM竞争性抑制剂,目前处于临床前研究阶段。它通过直接与酶结合并与甲基供体SAM竞争来抑制EZH2酶活性,该化合物对EZH2野生型和Y641F突变型均表现出浓度依赖性的抑制作用,IC50分别为(15f2)和(13f3)nmol · L-1。研究发现EL1处理的细胞显示H3K27me2和H3K27me3特异性减少且EZH1和EZH2基因敲除对H3K27甲基化和增殖的影响相似,下调PRC2复合物(如EZH2、EED和SUZ12)的siRNA可破坏该复合物并阻断肿瘤细胞的增殖,EL1在不破坏PRC2染色质结合的情况下抑制EZH2的HMT活性可以阻止肿瘤细胞的增殖和集落形成。抑制EZH2可能诱导生长停滞和向记忆B细胞分化。该研究首次证明了组蛋白H3K27me3的抑制可以在EZH2复合物与其靶基因不解离的情况下发生[49]。
2.2.11 HM-97594 HM-97594是韩美(中国)有限公司研发的一款小分子EZH1/2双重抑制剂,可用于治疗实体瘤和血液瘤,目前处于临床前研究阶段。体外研究显示,HM-9759对野生型EZH1的IC50为 2.8 nmol · L-1;对野生型 EZH2 的 IC50为 1.7 nmol · L-1;对突变型 EZH2(Y641F)的 IC50为 1.3 nmol · L-1。2019年12月,在美国佛罗里达州奥兰多举行的第61届美国血液学会(American Society of Hematology,ASH)会议和博览会上公布了体内研究结果:在美国癌症研究所(Institute of Cancer Researcch,ICR)小鼠模型中,给予重复剂量的HM-97594(50 mg · kg-1,qd),小鼠体质量、血液参数以及异常临床体征方面均无明显变化。在携带有EZH2 GOF突变的KARPAS-422(sc)异种移植肿瘤的 NOD SCID 小鼠中,HM-97594(25 mg · kg-1,PO,qd)治疗21 d后,肿瘤生长受到明显抑制,而体质量没有减轻。
2.2.12 DZNep DZNep是SAH水解酶的抑制剂,通过阻断在DNA甲基化过程中起重要作用的酶S-腺囊肌细胞水解酶(Ahcy)间接抑制EZH2[50]。目前正处于临床前研究阶段,DZNep已被证明能促进各种原发肿瘤细胞和癌细胞株的凋亡[51-56]。应用DZNep介导的凋亡效应在癌细胞中更为明显,对正常细胞的影响较小,并通过抑制H3K27me3标记[51,54,57]来促进凋亡。
3 结语与展望
EZH2对癌症有明显的功能增强作用和直接抑制肿瘤细胞内酶功能的能力,这使其成为了抗肿瘤治疗的一个引人注目的靶点,EZH2通过功能的获得、功能的丧失或过度表达从而导致肿瘤的发生。全球首个EZH2抑制剂tzemetostat的上市及其良好的安全性和有效性,大大激发了人们对EZH2这一高度成熟靶点的研发热情,本文总结了全球已上市和在研的11款EZH2抑制剂研究进展。相对于在恶性血液肿瘤中表现出的高度活性,EZH2抑制剂仅仅对少数具有SWI/SNF成员基因突变或者BAP1突变的实体瘤表现出一定的效果,在大多实体瘤中EZH2以过表达的野生型形式存在,同样具有H3K27me的催化活性。然而,目前尚不清楚过表达的野生型EZH2是否也是癌症依赖基因。因此,深入探究肿瘤细胞对EZH2抑制剂不敏感的分子机制及耐药机制是增强EZH2抑制剂在实体瘤中治疗效果的关键,也对新一代EZH2的开发具有指导意义。
猜你喜欢
中国农业科学(2022年14期)2022-07-26
传染病信息(2022年3期)2022-07-15
保健与生活(2022年11期)2022-06-09
新医学(2022年4期)2022-04-23
中草药(2022年1期)2022-01-13
海外星云(2021年9期)2021-10-14
现代临床医学(2021年4期)2021-07-31
现代仪器与医疗(2021年1期)2021-06-09
保健与生活(2021年5期)2021-04-12
大众健康(2020年7期)2020-08-25
