
双特异性抗体的药动学研究进展
2021-12-14 06:43曹庆娟张亚宁李甜甜杜娟娟
药学进展 2021年9期
曹庆娟,张亚宁,李甜甜,杜娟娟
(清华大学药学院,生命有机磷化学教育部重点实验室,北京 100084)
双特异性抗体(bispecific antibody,BsAb)是指通过基因工程改造,能同时结合同一个或不同分子上的2个不同抗原表位的抗体。这种抗体分子因具有特异的“双靶向”功能,能和2个不同的表位或抗原结合,发挥同时结合2个靶点的叠加或协同作用,甚至可以创造新的基本治疗作用机制[1-5]。抗体工程的发展促进了大量新型BsAb的出现,彻底改革了用于药物和临床诊断的BsAb开发的思路,使研究人员能够依照需求来调节BsAb的大小、价态、柔性、半衰期和生物分布等[6-8]。
截至2021年8月,全球有百余种BsAb正处于临床试验阶段,并已有4种药物上市,分别为卡妥索单抗(catumaxomab)、博纳吐单抗(blinatumomab)、艾美赛珠单抗(emicizumab)和埃万妥单抗(amivantamab)。卡妥索单抗是2009年由欧盟药品监督管理局(European Medicines Agency,EMA)批准在欧洲上市的小鼠和大鼠嵌合的免疫球蛋白G(immunoglobulin G,IgG)样BsAb,分别靶向T细胞上的分化簇3(cluster of differentiation 3,CD3)和上皮细胞黏附分子(epithelial cell adhesion molecule,EpCAM),用于治疗恶性腹水。鼠源抗体在人体内产生了较强的人抗鼠抗体反应和严重的不良反应(包括恶心、感染、腹部疼痛、肝功能异常等)[9-10]。由于上市后表现欠佳,卡妥索单抗曾于2017年停产退市。2020年6月,国家药品监督管理局药品审评中心批准了卡妥索单抗的临床试验(CTR20201246),适应证为伴腹膜转移的晚期胃癌,卡妥索单抗重新进入了审评程序。博纳吐单抗是2014年12月由美国FDA批准上市的双特异性T细胞衔接器(bispecific T cell engager,BiTE),相对分子质量约为55 000,由2个不同的单链可变区(single-chain variable fragment,scFv)通过柔性多肽串联而成,分别靶向T细胞的CD19和CD3,用于治疗B前体急性淋巴细胞白血病[11-13]。艾美赛珠单抗是2017年11月由FDA批准上市的IgG样BsAb,2个不同的抗原结合臂分别结合凝血因子IX/IXa和X/Xa,并模拟凝血因子VⅢ的部分功能,治疗A型血友病[14-16]。埃万妥单抗是2021年5月由FDA批准在美国上市的IgG样BsAb,分别靶向表皮生长因子受体(epidermal growth factor receptor,EGFR)和细胞间质上皮转换因子(cellular-mesenchymal epithelial transition factor,cMet),用于治疗EGFR突变的非小细胞肺癌[17-18]。
BsAb的 药 动 学 (pharmacokinetics,PK) 特征与BsAb的构建模式、相对分子质量、物理化学性质、靶标抗原以及和抗体可结晶段(fragment crystallizable,Fc)受体的相互作用等因素相关。BsAb的PK为临床研究中的给药方案和初始安全剂量提供参考。不同构建模式的BsAb在机体内的作用机制不同,其PK差异也较大。本文从BsAb的不同构建模式出发,详细比较了其PK差异,这为深入了解BsAb在机体内的作用机制,利用PK研究来指导和促进药物开发提供了理论基础。
1 目前在研的双特异性抗体及其结构
随着BsAb受到越来越多的关注,学术界和工业界发明了多种构建BsAb的方法。目前,已有超过100种BsAb的构建模式。采用这些构建模式所获得的BsAb结构多样性大、相对分子质量范围广、蛋白质的理化性质差异显著,因此,BsAb的结构对于其PK性质有着重要的影响。因为BsAb构建模式种类繁杂,本文将重点集中在目前已经进入临床试验阶段的BsAb,关注它们的PK表现。截至2021年8月,除已上市的BsAb外,在研的BsAb有百余种处于临床试验阶段(见表1、2)[19]。这些抗体主要由25种构建模式构成,其相对分子质量分布于25 000 ~ 1 000 000不等,对于2个抗原的结合价态也有1+1、1+2和2+2等不同模式。为了后续更好地讨论PK,本文将这些BsAb的构建模式分为不含Fc区的BsAb和含Fc区的BsAb 2类(见图1)。
不含Fc区的BsAb的相对分子质量较低,均低于单克隆抗体(monoclonal antibody,mAb)药物,其结构也与IgG的结构差异较大(见表1和图1)。这类BsAb主要包括以下几种:由纳米抗体(nanobody,Nb),即重链单域抗体(variable domain of heavy chain antibody,VHH)串联而成的串联纳米抗体(tandem VHH,TdVHH)[20]、scFv串联而成的BiTE、单链双价抗体(single-chain diabody,scDb)、抗体的重链可变区(variable domain of heavy chain,VH)和轻链可变区(variable domain of light chain,VL)分别串联的双亲和重定向抗体(dual affinity re-targeting,DART)[21]、scFv和 抗原结合片段(fragment antigen binding,Fab)串联而成的scFv-Fab和(scFv)2-Fab、四价的串联双抗体(tandem diabody,TandAb)。

表1 处于临床试验及已上市的不含Fc区的双特异性抗体Table 1 BsAbs without Fc region under clinical trials or on the market
相比于不含Fc的BsAb,含有Fc的BsAb的相对分子质量较大,多数结构与IgG更为相似;其中有部分具有IgG经典的Y型结构以及相似的相对分子质量,这部分BsAb也被称为IgG样BsAb(IgG-like bsAb)。这类BsAb主要包括以下几种(见表2和图1):在铰链区插入scFv的Fab-scFv-Fc、在轻链的C端串联多肽的IgG-peptide、Fc区C端融合scFv的IgG-(scFv)2[22-26]、Fc区两端均连接scFv的(scFv)4-Fc、Fc异二聚体且其中1个Fab臂的CH1和CL区互换形成的CrossMab抗体、CrossMab一臂串联1个Fab的F(ab)3CrossMab[27-29]、Fc区连接DART的(DART)2-Fc、Fc异二聚体且轻重链正交配对形成的 DART-Fc和LP-DART[30-31]、通过突变构建Fc异二聚化及轻重链正交配对(hetero H-ortho HL)、串联纳米抗体和Fc融合的TdVHH-Fc、IgG的N端串联第2个可变区的双特异四价IgG样抗体(dual-specific tetravalent IgG-like,DVD-Ig)[32-33]、Fc形成异二聚体并使用共同轻链的Hetero H-cLIgG、Fc异二聚体控制的Fab臂重组抗体Hetero H-Fab-arm exchange IgG、2个带有单链Fc的BiTE(BiTE-Fc)、IgG的2个抗原结合臂分别为Fab和scFv的Hetero H-scFv/Fab、突变Fc以结合抗原的mAb2[34]、通过纯化κ/λ轻链获得的κ/λ抗体(purified κ/λ IgG)[35]、两臂分别为 TdVHH和 Fab的 Fc异二聚体(hetero H,TdVHH/Fab)[36]、交叉双可变Ig样 抗 体 (cross-over dual variable Ig-like proteins,CODV-Ig)[37-38]、Fab交叉串联抗体(Fabs-in-tandem immunoglobulin,FIT-Ig)[39]、IgM 与 scFv融 合 的IgM-scFv 抗体[40]。
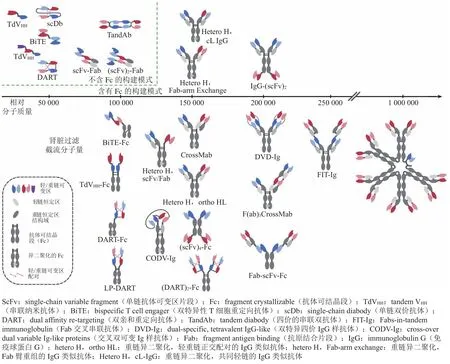
图1 部分处于临床试验阶段及已经上市的双特异性抗体的构建模式Figure 1 Construction formats of some bispecific antibodies under clinical trials or on the market
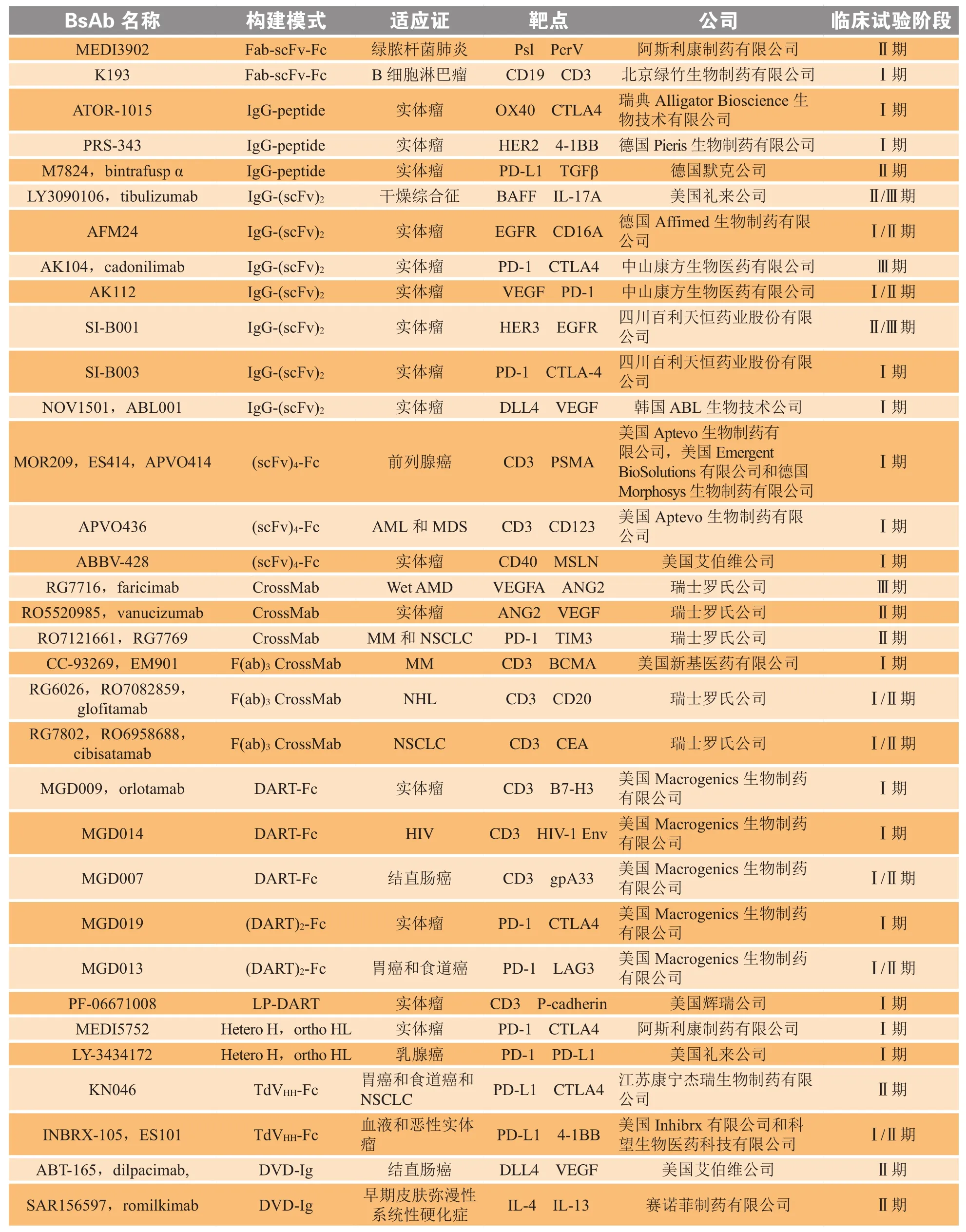
表2 处于临床试验及已上市的含Fc区的双特异性抗体Table 2 BsAbs with Fc region under clinical trials or on the market

续表2
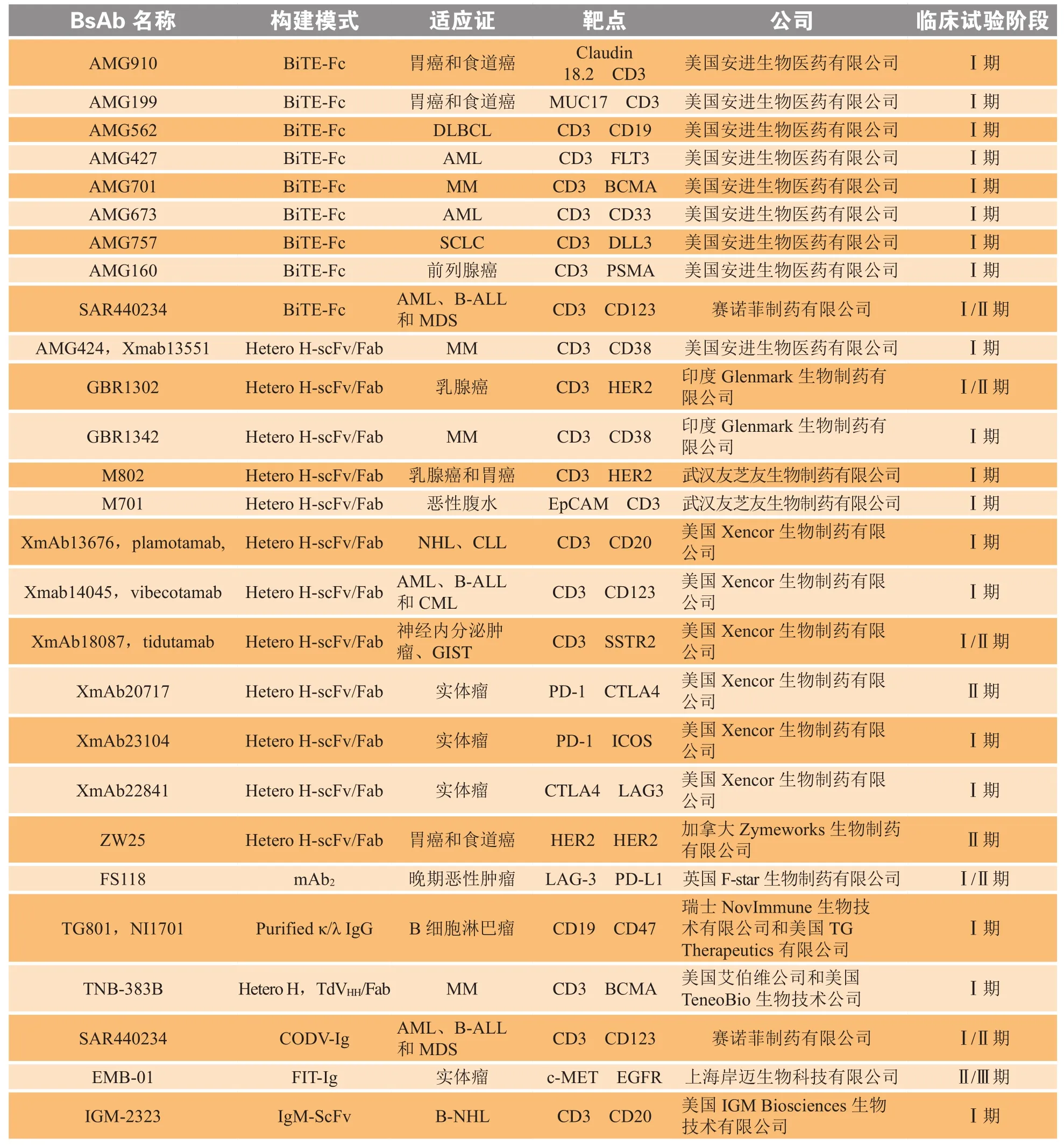
续表2
2 双特异抗体的药动学研究
BsAb在体内的药动学过程相比于小分子以及单靶点的其它蛋白药物都更为复杂。同时,BsAb的相对分子质量分布范围广、结构差异性大、构建模式多样性高;因此,相比于单抗药物,影响BsAb药动学的因素更加复杂多样。
2.1 吸收
BsAb作为治疗性蛋白药物,在人体的吸收中具有蛋白类药物,特别是经典的单克隆抗体药物的特点。目前BsAb主要以静脉注射的方式给药,这能使BsAb快速地在系统循环中分布并达到较高浓度[41]。除了静脉注射,BsAb也可在皮下或肌肉注射后吸收。参考单抗药物和其他蛋白药物,这些大分子蛋白(相对分子质量> 20 000)在皮下或者肌肉注射后,主要随着组织间隙液体的对流进入淋巴系统,再从淋巴系统缓慢引流进入血液循环。这一吸收过程往往持续数小时[42]。相对于单抗药物,对于BsAb经皮下和肌肉注射给药途径后的吸收情况的报道仍较少。AMG212(构建模式:BiTE,靶点:PSMA×CD3)在小鼠皮下注射2 h后,血浆药物浓度达到峰值,即血药峰浓度(peak concentration,Cmax),生物利用度为 18%[42]。 AFM11(TandAb,CD3×CD19)经皮下给药8 h后在小鼠体内达到血药峰浓度,生物利用度约为24%[43]。上述2种BsAb的生物利用度较单抗药物(生物利用度约50% ~100%)更低,可能与吸收前的药物降解有关。影响吸收前降解的因素有很多,包括组织内蛋白酶解、注射部位的内吞作用和新生儿Fc受体(neonatal Fc receptor,FcRn)介导的回收[44]。在一个48位健康志愿者参与的试验中,采用抗体样结构的伊美珠单抗(emicizumab)(Hetero H-cL-IgG, FIXa×FX/FXa)经皮下注射后,平均6.9 ~ 11.4 h后血药浓度达到峰值,平均生物利用度可达80% ~ 93%[45];这一较高的生物利用度可能部分来自FcRn介导的BsAb的回收。
2.2 分布
类似单抗药物,BsAb从血液进入组织主要是跟随体液的流动,而进入淋巴系统的BsAb部分再次回流进入血液。携带Fc区的BsAb还能通过FcRn介导的胞吞转运,从血管外渗并再循环到血管中[41]。BsAb的构建模式、细胞旁路运动(如对流、扩散)、FcRn介导的胞吞转运、靶标结合和内吞、非特异性代谢等多个因素都影响BsAb在机体内的分布。
虽然不同BsAb的相对分子质量和结构都有较大的差异,它们的分布容积却较为类似。例如:伐 努 赛 珠 单 抗(vanucizumab)(CrossMab,ANG2×VEGF)在人体内的稳态分布容积为2.9~ 6.1 L, 和 单 抗 相 似(3 ~ 5 L)[46];MEDI3902(Fab-scFv-Fc,Psl×PcrV)在人体内的稳态分布容积为3.4 ~ 4.9 L[47]。相对分子质量较小的博纳吐单 抗(blinatumomab)(BiTE,CD3×CD19) 在人体内的稳态分布容积为3.4 L,约等于人的血浆容积,与伐努赛珠单抗和MEDI3902类似[48]。这些结果说明了BsAb的分布途径相似,均为跟随体液流动,而分布范围也大多局限在血液和灌流较好的器官。
另外,与单抗药物类似,BsAb药物的PK也可以采用两室模型进行较好的拟合[49-52],为深入理解BsAb的体内分布提供了更多的信息。例如,那赛昔珠单抗(navicixizumab)(Hetero H-cL-IgG,DLL4×VEGF)中央室的分布容积为 41.6 mL · kg-1,接近人生理血浆容量(40 ~ 45 mL · kg-1),外周室的分布容积为26.4 mL · kg-1,表明那赛昔珠单抗主要分布在血管中,中等程度地溢出到周围组织,符合经典 IgG 的特性[50]。PF-06671008(LP-DART,CD3×P-cadherin)在人体中的稳态分布容积为251 mL · kg-1,中央室分布容积为 40.2 mL · kg-1,外周室分布容积为211 mL · kg-1[51]。相似结构的MGD007(DART-Fc,CD3×gpA33)在人体内的稳态分布容积为 123 mL · kg-1,中央室分布容积为 51.3 mL · kg-1,外周室分布容积为72 mL · kg-1[21]。这2种BsAb的外周室分布的差异,可能主要来源于对于靶点胎盘型钙黏蛋白(placental cadherin,P-cadherin)或者糖蛋白A33(glycoprotein A33,gpA33)的结合。
在其他结构类型的BsAb中,也观察到了分布容积与其靶点相关的现象。例如:同为BiTE结构的AMG212(BiTE,PSMA×CD3)和AMG596(BiTE,EGFRvIII×CD3)在小鼠静脉注射后的稳态分布容积分别为1 000和160 mL · kg-1,其差异可能也来自于靶点介导的分布。同样靶向PSMA的MOR209[(scFv)4-Fc,CD3×PSMA]在小鼠中也显示了较高的分布容积760 mL · kg-1,佐证了上述PSMA靶点介导的分布现象[53]。
2.3 清除
类似单抗药物,BsAb也可通过排泄或代谢途径从体内清除。BsAb的清除速率与其半衰期、人源化程度、与FcRn的亲和力、免疫原性、糖基化的性质和程度以及对蛋白水解酶的敏感程度等相关[54-55]。BsAb从体内清除还受非特异性代谢、肾脏清除、靶标介导的清除、FcRn介导的回收、抗药抗体(anti-drug antibody,ADA)的产生等因素影响。
2.3.1 非特异性清除 非特异性代谢广泛发生在整个机体内,BsAb通过胞饮作用进入细胞而被降解[48]。胞饮作用是细胞中一种相对非特异性的液体内吞作用。胞饮摄取后的药物蛋白的分解代谢降解不局限于某个特定的器官,而是遍布全身,特别是在富含毛细血管床的器官和组织中。因此,皮肤、肌肉和胃肠道是通过胞饮作用清除蛋白药物的主要清除器官。因为血管内的内皮细胞的总表面积大(总表面积> 1 000 m2),该过程可以有效地将蛋白药物分子从体内清除,是BsAb清除的主要途径之一。
2.3.2 肾脏清除 肾脏清除和BsAb的相对分子质量相关[56]。一般认为,肾脏对于球状蛋白的截留相对分子质量约为60 000(见图1)。相对分子质量小于这一值的BsAb主要经由肾脏清除,半衰期较短。例如:BiTE构建模式(相对分子质量为55 000)的博纳吐单抗(BiTE,CD3×CD19)在人体中半衰期 为 2.1 h[57];solitomab(BiTE,CD3×EpCAM)在人体内半衰期约为4.5 h[58];AMG212(BiTE,PSMA×CD3)在小鼠中的半衰期约8 h[42];AMG211(BiTE,CD3×CEA)在小鼠中半衰期为2.2 ~ 6.5 h[42]。DART构建模式(相对分子质量约59 000)的对氟替珠单抗(flotetuzumab)(DART,CD3×CD123)在食蟹猴中的半衰期为3.5 h[59]。由于这些BsAb的半衰期短,往往给药频率高,或者采用连续输液的方式给药。
超过肾脏截流相对分子质量的BsAb,极少通过肾脏过滤排出,半衰期较长[60]。例如TandAb构建模式(相对分子质量约105 000)的AFM13(TandAb,CD30×CD16A)在人体内半衰期为8.72 ~ 19.2 h[61];AFM11(TandAb,CD3×CD19)在小鼠中半衰期为 18 ~ 22 h,清除速率为 0.5 mL · h-1· kg-1,小于小鼠的肝脏和肾脏的血流速率,表明肾小球过滤和肝脏降解都未主导AFM11的清除[43]。
2.3.3 靶标介导的清除 当BsAb靶向细胞膜受体时,BsAb也可通过特异性靶标介导的途径进行清除。当BsAb浓度低于靶受体时,靶标介导的清除途径未饱和,BsAb清除速率呈线性,速率高;当BsAb浓度高于靶受体时,靶标介导的清除途径饱和,BsAb清除速率变慢,呈非线性[41]。例如dilpacimab(DVD-Ig,DLL4×VEGF)在食蟹猴中经静脉注射后显示出非线性的PK特征:当高剂量(注射剂量≥10 mg · kg-1)时,PK参数接近线性范围,类似典型的单抗,清除速率较低(清除速率≤ 5.1 mL · d-1· kg-1),半衰期 > 5 d;而在低剂量时,则具有更高的清除速率和更短的半衰期[32]。非线性清除可能是由于在高浓度下,细胞膜靶标DLL4介导的dilpacimab清除已经达到饱和。那赛昔珠单抗(navicixizumab)(Hetero H- cL-IgG,DLL4×VEGF)在低剂量(注射浓度为 0.5 mg · kg-1)组和中剂量(注射浓度为1.0 mg · kg-1)组的平均清除速率分别为 6.24 和 6.45 mL · d-1· kg-1,比高剂量组(注射浓度为2.5 mg · kg-1)或更高剂量注射组的清除速率稍高,表明存在中等程度的靶标介导的清除[50]。对氟替珠单抗(DART,CD3×CD123)也通过2种靶标受体CD3和CD123介导的清除途径导致药效减弱[59]。
2.3.4 FcRn介导的回收对于双特异性抗体清除的影响 FcRn可以通过再循环作用将内吞进入细胞的抗体或者血清白蛋白回收进入血液循环。因此,这一途径使带有Fc的BsAb具有更低的清除速率。例如:BiTE-Fc(相对分子质量约100 000)构建模式的 APVO425(BiTE-Fc,ROR1×CD3)在小鼠中的半衰期为 6.25 d[52],AMG701(BiTE-Fc,CD3×BCMA)在食蟹猴中的半衰期为4.7 d[62],AMG673(BiTE-Fc,CD3×CD33) 在 人 体 内 的半衰期为1.8 ~ 7 d[63]。(scFv)4-Fc(相对分子质量约150 000)构建模式的MOR209 [(scFv)4-Fc,CD3×PSMA]在小鼠中的半衰期为4 d[53]。FabscFv-Fc构建模式(相对分子质量约200 000)的MEDI3902(Fab-scFv-Fc,Psl×PcrV)在人体内的半衰期为7.2 ~ 8.4 d[47]。
对Fc进行突变从而促进Fc异二聚化是构建BsAb的一类重要方法。与野生型Fc类似,这些突变的Fc也可被FcRn回收。例如:Hetero H-scFv/Fab构建模式的M802(Hetero H-scFv/Fab,CD3×HER2)在小鼠中的半衰期为2.7 d[64];Hetero H-scFv/Fab构建模式的Xmab14045(Hetero H-scFv/Fab,CD3×CD123)在小鼠中半衰期为 6.2 d;XmAb13676(Hetero H-scFv/Fab,CD3×CD20) 在小鼠中的半衰期为6.7 d[65]。LP-DART构建模式的PF-06671008(LP-DART,CD3×P-cadherin) 在 小鼠中的清除速率为 4.6 mL · h-1· kg-1,由于 Fc 区存在,其半衰期相比于DART模式的类似抗体延长至4 d[51];MGD007(DART-Fc,gpA33×CD3)在食蟹猴中的清除速率为 0.7~0.8 mL · h-1· kg-1,半衰期为 6.1 ~6.8 d[21]。
Fc异二聚化使人们可以构建结构与野生型IgG更为相似的IgG样BsAb,这些BsAb往往具有更接近单抗的PK表现。例如:CrossMab构建模式的 伐 努 赛 珠 单 抗 (vanucizumab)(CrossMab,ANG2×VEGF)在人体内的血液清除速率为10.9 ~36.7 mL · h-1· kg-1,和单抗类似,半衰期为 6 ~ 9 d[46]。Hetero H-cL-IgG构建模式的tepoditamab(Hetero H-cL-IgG,CD3×CLEC12A)在人体内的半衰期为 9.4 d[66];REGN4018(Hetero H-cL-IgG,CD3×MUC16)在食蟹猴中的半衰期约10 d[67];那赛昔珠单抗(Hetero H-cL-IgG,DLL4×VEGF)在人体内的半衰期为11.4 d[50]。
除了Fc外,血清白蛋白(human serum albumin,HSA)也可以通过FcRn回收,沃巴利珠单抗(vobarilizumab)(TdVHH,IL-6R×HSA)即通过结合 HSA,降低清除速率到 0.38 ~ 1 mL · h-1· kg-1,在食蟹猴中半衰期从4.3 h延长至1.7 ~ 6.6 d[68]。
2.3.5 双特异性抗体特殊的清除机制 多项研究中,均观察到了含有Fc的BsAb的体内循环时间短于野生型的IgG,说明其相比于IgG有更高的清除速率[69-71]。Datta-Mannan等[72]发现双特异性IgG-scFv或IgG-ECD抗体在食蟹猴中出现了野生型IgG和Fc融合蛋白都不存在的一种代谢途径,导致了BsAb的循环时间显著低于IgG。经过研究证明,这个特殊的清除机制是通过肝脏中肝窦内皮细胞(liver sinusoidal endothelial cell,LSEC)进行的;并且,这种BsAb的快速清除并非归因于受体介导的内吞、Fc-FcRn相互作用的降低或BsAb的理化性质的不同。2019年,Datta-Mannan等[73]又发现这一特殊的清除过程不仅存在于肝脏中,肾脏和脾脏中也存在相似的过程。同时,当抗体上融合的胞外结构域(extracellular domain,ECD)的位点位于重链的C端时,这一过程更加显著;而当ECD的融合位点位于重链N端时,则这一清除机制大大降低。目前,这一特殊清除机制的具体机理尚未完全研究清楚,可能和BsAb在体内环境中的构象改变有关。
2.3.6 抗药抗体对于双特异性抗体清除速率的影响 抗药抗体(anti-drug antibody,ADA)的产生是人体免疫系统将药物识别为外源蛋白后产生的免疫反应,可能对BsAb的清除速率产生影响从而改变BsAb的PK表现,有些ADA可以中和BsAb消除其生物功能。因此,ADA的产生是影响BsAb的PK-药效学(pharmacodynamics,PD)的重要因素。早期非人源的BsAb(如catumaxomab),会引起较强的ADA反应[74]。随着人源化抗体和全人源抗体的推广,虽然治疗性抗体(包括单抗和BsAb)的免疫原性有很大改善,但这一问题却仍然伴随着抗体类药物的开发和使用。
相比于传统单抗药物,BsAb的结构或者序列与内源抗体存在较大差异,更容易产生抗药抗体。在多个BsAb的临床试验中均观察到ADA的产生,其中一部分ADA对药物暴露和抗体清除速率无影响,例如伐努赛珠单抗(CrossMab,ANG2×VEGF)给药后约5%病人产生ADA,但药物暴露量未发生变化[46]。Ozoralizumab(TdVHH,TNFα×HSA×HSA)使2.6%病人呈ADA阳性,但ADA对药物暴露量和清除速率均无影响[75]。另外,有些BsAb的临床试验中出现了影响药物PK的ADA。在那赛昔珠单抗(Hetero H-cL-IgG,DLL4×VEGF)治疗的患者中,约半数ADA阳性的患者提高了那赛昔珠单抗的清除速率,导致了药物暴露量降低[76]。在emicizumab(Hetero H-cL-IgG,FIXa×FX/FXa)的一项试验中,6.7%的患者产生了ADA,并且其中3人产生的ADA影响了药物的PK[77]。在AMG211(BiTE,CD3×CEA)的临床试验中,观察到48.7%的病人产生了ADA,其中部分病人产生的ADA降低了药物的浓度[78]。更强的抗药免疫反应可以导致机体针对BsAb产生中和性抗药抗体(neutralizing ADA)。例如,AFM13(TandAb,CD30×CD16A)在54%病人中产生ADA,其中29%病人产生的ADA具有中和能力,低剂量(药物注射浓度为0.15 mg · kg-1)和高剂量(药物注射浓度为1.5 mg · kg-1)组均有高水平的ADA产生,导致药效减弱[61]。博纳吐单抗(BiTE,CD3×CD19)导致少数约0.9%病人产生了中和性ADA[48]。在注射ozoralizumab(TdVHHs,TNFα×HSA×HSA)的病人中,2.6%病人也产生了中和性ADA[75]。总之,BsAb可以产生不同程度的免疫原性,但总体来说,没有研究报道BsAb的ADA会引起非常严重的不良反应。
BsAb的免疫反应和很多因素相关。与单抗不同,BsAb是人造的蛋白结构;构建BsAb时采用的突变、融合等方法均可能在蛋白上引入产生免疫原性的位点。此外,蛋白的聚集对于蛋白的免疫原性有很大的影响[75],而不同构建形式的BsAb的聚集程度不同,会显著影响其ADA的产生。并且Fc可以提高蛋白的免疫耐受[79]。因此,BsAb的免疫原性和其构建模式相关。
另外,给药剂量、途径、频率、患者的疾病状态等均会影响BsAb的免疫原性[80]。例如,那赛昔 珠 单 抗 (Hetero H-cL-IgG,DLL4×VEGF) 低剂量水平(药物注射浓度≤2.5 mg · kg-1)给药时,约56%患者呈ADA阳性;而高剂量水平(药物注射浓度为3.5 ~ 5 mg · kg-1)给药时,仅11%患者产生ADA[76]。BsAb的免疫原性往往也受给药途径的影响,从高到低的免疫原性等级依次为皮下注射、肌肉注射以及静脉注射[76]。ADA对药物暴露和药效的影响也和病人相关,例如AMG211(BiTE,CD3×CEA)在48.7%病人中产生ADA,其中12.8%病人体内的ADA水平较高;7.7%病人中高水平ADA未影响药物暴露量;而5.1%病人的高水平ADA引起药物浓度下降,药效降低[81]。
3 基于机制的药动学/药效学模型在双特异性抗体药物开发中的应用
与单纯的PK不同, PK-PD模型将PK和PD模型集成到一套数学模型中,从而描述给药后药物暴露、效应与实践的动态关系。PK-PD模型可以用于优化药物剂量、优化临床试验的设计以及指导药物的设计等方面。近年来,PK-PD模型已经从传统的经验性的模型过渡到基于机制的PK-PD模型,后者基于机体的生理/病理特征或药物作用机制构建,具有更高的预测准确度[82-85]。随着BsAb药物的发展,基于机制的PK-PD模型也逐步被用以模拟BsAb的PK特征和组织分布。例如:Glassman等[82]利用靶点介导的药物处置模型(target-mediated drug disposition,TMDD)模拟 2种BsAb的PK和PD表现,同时,也模拟了不同亲和性的抗体的PK和PD表现的不同,为BsAb的设计提供了参考。
T细胞招募的BsAb通过BsAb将T细胞和肿瘤细胞进行交联,诱导形成三元免疫复合体,从而激活T细胞杀伤癌细胞,是重要的BsAb类型。多个课题组对于这类BsAb进行了基于机制的PK-PD模型模拟。Schropp等[86]则采用TMDD模型,建立了T细胞招募BsAb的PK-PD模型,模拟了三元免疫复合物的产生和BsAb剂量的关系,为BsAb计量的选择提供了参考。Campagne等[59]采用基于机制的PK-PD模型,研究非人类灵长类动物中DART模式的BsAb fotetuzumab(DART,CD3×CD123),描述了DART分子与外周血CD3表达细胞和CD123阳性细胞的结合、T细胞的激活和增殖,以及由此导致的外周血CD123细胞的清除。Betts等[51]采用定量系统药理学的方法,提出基于机制的PK-PD模型,描述小鼠药效模型中BsAb PF-06671008(LPDART,CD3×P-cadherin)的体内PK-PD关系。该模型可用于预测临床上的PK-PD参数,并为药物开发不同阶段的药物设计、亲和力优化、用药剂量等的设计提供参考[51]。
基于机制的PK-PD模型对于首次人体试验(first in human,FIH)药物剂量的选择可以提供重要的参考价值。在对BsAb PF-06671008(LP-DART,CD3×P-cadherin)的研究中,Chen等[87]建立了一个基于机制的PK-PD模型,用以阐释剂量-药效的关系,从而帮助推算FIH的药物剂量。Sacks等[88]基于PK模型和免疫突触形成机制,建立了整合PK-PD模型,并结合灵长类临床前试验数据,预测临床PK-PD特征,给出了用于FIH研究的推荐药物剂量[88]。
4 结语
BsAb是一类新型的抗体类药物,其作用机制新颖多样,为多种疾病提供了新型的治疗策略。设计BsAb药物需要考虑多种因素,如对靶标的亲和力、选择性、可能的作用机制等,同时PK特性也有可能会显著影响BsAb的药效。因此,了解BsAb的PK特性和影响其吸收、分布和清除的重要因素是BsAb药物开发和临床试验的关键。然而,BsAb构建模式多样,往往对于抗体结构或序列做较大的改造,导致其PK表现更复杂。BsAb的PK特性与经典的单抗有一定的相似性,例如,其吸收和分布的特性都与单抗相似。另一方面,BsAb的PK也高度依赖于其结构和构建模式。不同构建模式的BsAb有着不同的结构、相对分子质量和Fc受体结合特性等,导致其PK特性存在明显的区别。例如,BsAb的清除速率就受其构建模式的显著影响。另外,由于BsAb是通过突变、融合等方式改造单抗来获得,也更容易产生ADA,并可能对其PK特性产生影响。目前,针对BsAb的PK研究仍然较少,相信随着BsAb应用的进一步推广,可以获得更多关于其PK的有效信息,为设计更好的BsAb提供指导。
猜你喜欢
实用肿瘤学杂志(2022年3期)2022-11-30
医院管理论坛(2022年7期)2022-10-14
现代仪器与医疗(2022年4期)2022-10-08
健康体检与管理(2022年4期)2022-05-13
实用肿瘤学杂志(2021年2期)2021-12-01
现代临床医学(2021年5期)2021-11-02
证券市场红周刊(2019年6期)2019-06-11
中国实用医药(2016年9期)2016-05-17
图书与情报(2015年2期)2015-11-21
中学化学(2014年11期)2015-01-20
