
铁铜双核羰基簇合物结构的理论研究
2021-12-27 09:13张建辉冷艳丽罗迎春王环江慕红梅
原子与分子物理学报 2021年6期
张建辉, 冷艳丽, 罗迎春, 王环江, 慕红梅
(1. 贵州民族大学 化学工程学院, 贵阳 550025; 2. 兰州资源环境职业技术学院 环境与化工学院, 兰州 730021; 3. 甘肃伯骊江3D打印科技有限公司, 兰州730021)
1 引 言
在非均相催化反应中, 催化剂常常由小金属团簇沉积在氧化物表面组成, 由于团簇的尺寸范围很宽, 不同尺寸的团簇催化活性也不相同, 研究团簇的催化活性有助于选择性地合成催化剂[1], 大量研究结果表明, 纳米金属簇因为独特的量子尺寸效应、体积效应以及表面效应, 导致其催化活性和选择性的飞跃, 被称为“第四代催化剂”. 双金属催化剂因为其独特的催化优点被广泛研究. 通过添加一定数量的其他金属对现有金属催化剂进行修饰是双金属催化的主要研究方法, 该修饰过程改变了现有金属催化剂的电子结构, 与单金属的催化相比双金属簇合物具有独特的催化行为[2,3]. 因过渡金属羰基配位化合物同时具备无机和有机化合物的特性, 关于该类化合物的研究是国际上化学研究中蓬勃发展的领域之一. 研究已从简单的单核配位化合物逐渐转向双核和多核配位化合物体系. 1890 年, Mond等人在光作用下通过过渡金属镍与一氧化碳直接合成第一个过渡金属羰基化合物Ni(CO)4[4]. 1905 年, Dewar等人利用 Fe(CO)5合成双核过渡金属羰基化合物 Fe2(CO)9[5]. 理论研究方面主要采用 DFT 等量子化学方法, 研究了第四周期过渡金属M2(CO)n在 16、17 和 18 电子数结构下的成键问题[6,7]. 其中铁是研究最多的过渡金属之一. 已报道的过渡金属双原子羰基配位化合物有 Cr2(CO)n(n=11-8)、Mn2(CO)n(n=10、9、8)、Fe2(CO)n(n=0-9)、Co2(CO)n(n=8、7、6)、Ni2(CO)n(n=7、6、5) 和 Cu2(CO)n(n=6-1)等[8-13]. 通过以上研究发现相比于单核配位, 双核或多核配位化合物的结构和键合方式复杂, 有特殊的成键和结构规律, 鉴于实验条件的限制, 从理论方法上对其物质结构和性质的预测有着十分重要的意义.
早在1926年费托(Fischer-Tropsch)[14]合成提出时, 就已经发现Cu助剂能降低Fe基催化剂的还原温度, 促进催化剂的还原, 后来的很多实验也验证了这一点, 因此Cu作为一种还原助剂, 经常用于费托(F-T)合成Fe基催化剂中.Pakiari[15]采用DFT理论研究了纯Fe团簇和Fe被Cu取代的FeCu合金团簇FenCum(2≤m+n≤4)对乙烯吸附性能的影响, 发现Cu 取代部分Fe 原子的合金团簇对乙烯的吸附更强, 且对乙烯与金属团簇的成键方式、键能等也有显著影响.由于F-T 反应过程中催化剂活性相的复杂性和中间物种的不稳定性, 使得F-T 反应机理十分复杂. 多年来, 研究人员提出了多种机理, 其中作为最初反应物的CO的吸附解离是研究各反应过程的关键. 本文将研究FeCu双原子团簇对CO的吸附过程, 得到团簇原子替代对的吸附规律影响, 为获得高活性及高选择性的催化剂提供理论依据.
2 计算方法
采用密度泛函理论BPW91方法[16,17]和6-311+G (d)基组[18], 对FeCu金属团簇在不同自旋态下的结构进行计算, 且不加任何对称性限制. 为了确保优化所得结构为势能面(PES)上的最低点, 在结构优化完成后进行频率计算(虚频数为零, Nimag=0), 根据能量最低原理确定FeCu团簇的基态. 在基态FeCu的结构基础上, 对可能的CO吸附结构全面计算, 以逐步得到CO吸附过程中配合物的最稳定基态结构. 全部计算工作采用Gaussian 09程序完成[19],分子的几何构型由Gauss View程序从计算结构直接转换而来.
吸附能计算采用以下公式完成:Ead=Etotal-E(FeCu)-nE(CO), 其中Etotal、E(FeCu)和E(CO)分别为:FeCo(CO)n、FeCu和 CO分子的能量,Ead值越负说明该反应放热越多.
3 结果与讨论
3.1 FeCu和FeCuCO的结构及其相关性质
FeCu团簇和 FeCuCO羰基配合物在不同多重度的相关信息和结构参数如表1和图1所示, 根据计算FeCu团簇的基态为四重态, 电子态为4∑, 比二重态和六重态能量分别低 100.8和37.1 kJ·mol-1, FeCu键长为2.278 Å. 各多重度下Mulliken电荷分布为:2Fe(0.221)-Cu(-0.221)、4Fe(0.205)-Cu(-0.205)、6Fe(0.233)-Cu(-0.233)、8Fe(0.073)-Cu(-0.073) 和10Fe(0.042)-Cu(-0.042). FeCuCO羰基配合物的基态为四重态, FeCu键长为2.315 Å, 比二重态和六重态能量分别低 81.3和117.3 kJ·mol-1, FeCu键长为2.278 Å.

图1 bpw91/6-311+G(2d)方法下FeCu 和FeCuCO各多重度的结构Fig. 1 Geometries of the FeCu and FeCuCO at the bpw91/6-311+G(2d) level on the states (bond length in angstrom and bond angle in degree)
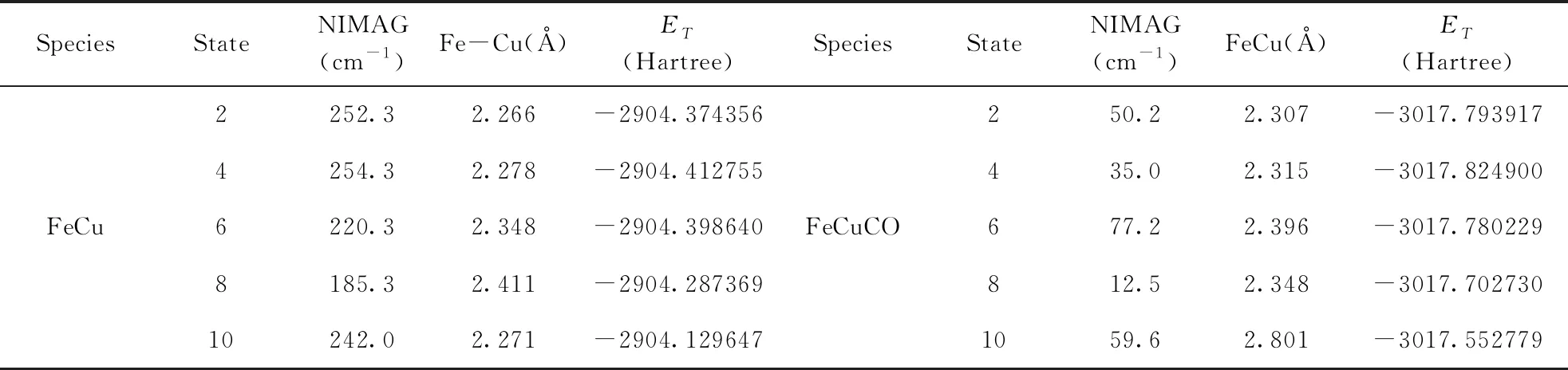
表1 FeCu团簇和 FeCuCO羰基配合物在各多重度的FeCu的键长(Å), 振动频率(cm-1)和体系总能量(hartree)
3.2 FeCu(CO)n羰基配合物的结构及其相关性质
FeCu(CO)n羰基配合物的结构参数和电子态的相关信息见图2, 吸附CO过程中的FeCu的键长、吸附能等性质如表2所示. 对于异核双原子团簇,首先考虑不同原子以及共同吸附对CO的作用能力, 通过对各吸附过程的计算发现, 第一个CO分子首先与FeCu的铁原子发生吸附, 基态为四重态, 电子态为4A". 第二分子CO的吸附位置是铁铜桥键, 基态为二重态, 电子态为2A′. 第三分子CO的吸附改变了第二分子CO的吸附位置, 此时三分子CO都吸附在铁原子上, 第四分子CO的吸附位置仍然是铁原子, 当第五分子CO发生吸附时, 吸附位置发生变化, 吸附在铜原子上, 主要原因是Fe(CO)4一侧已经达到18电子, 不可能继续对CO进行吸附. 从变化过程我们发现, 吸附过程中结构的渐变是Fe原子先达到18电子规则生成 Fe(CO)4Cu, 后续的吸附过程是在保持Fe原子18电子规则的基础上, 在Cu原子上发生吸附. 第六分子CO吸附类型是铁铜桥式羰基, 同时吸附在铁原子上第四分子CO改变为吸附到铁铜桥键上. 第七分子CO的吸附位置是铜原子, 但改变了第六分子吸附时的结构, 同时第四分子的CO恢复了铁原子配位, 第八分子CO吸附热为1.9 kJ·mol-1, 说明第八分子CO不易吸附. 整个吸附过程中体系的多重度从Fe(CO)2Cu后均为二重态. Fe(CO)1Cu的生成反应吸附能最大, 放热199.0 kJ·mol-1.
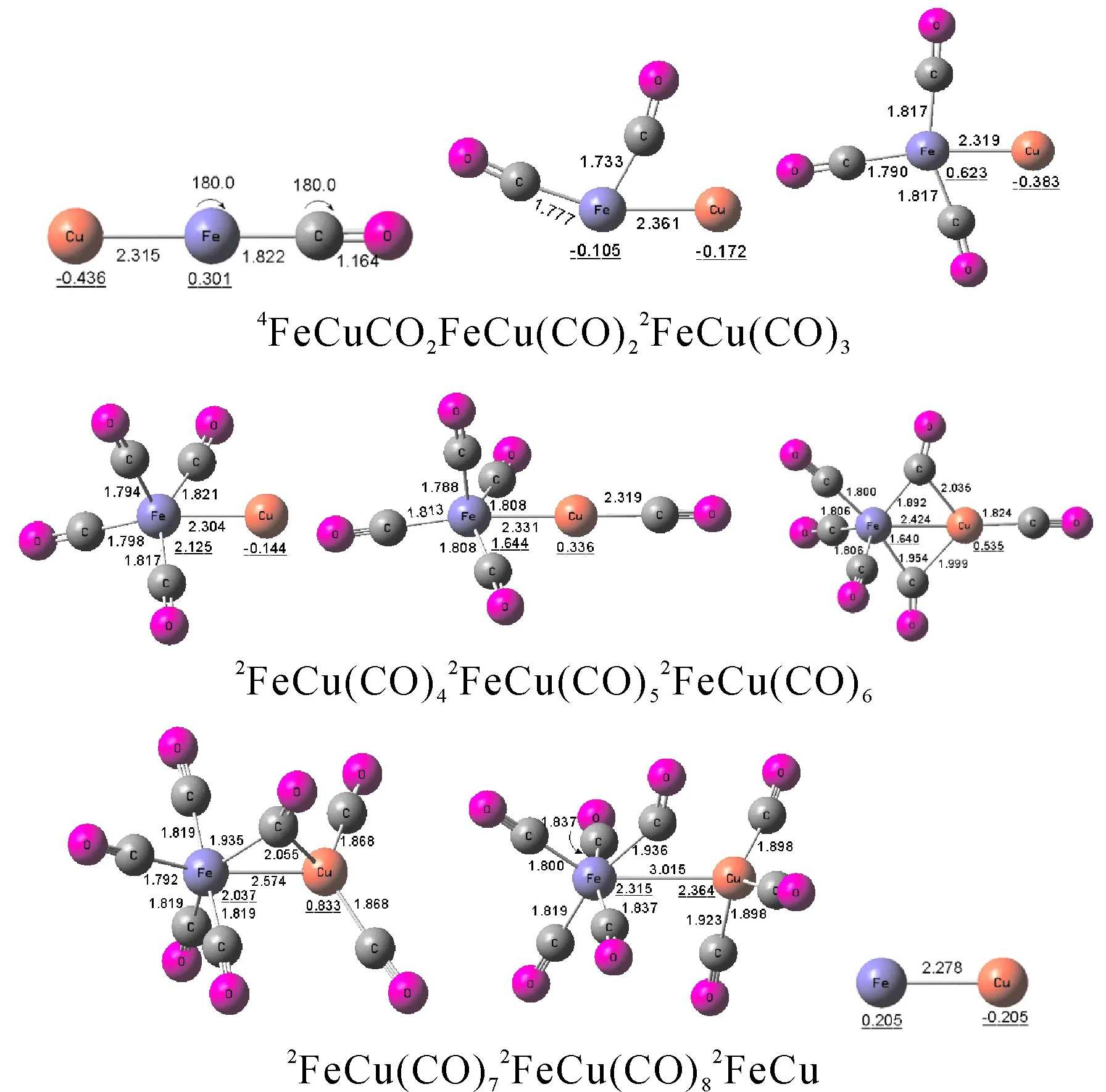
图 2 FeCu(CO)n (n=1-8)的基态结构和电荷密度Fig. 2 Structures and spin densities (underlined) of the FeCu(CO)n (n=1-8) on the ground state

图 3 FeCu(CO)n (n=1-8)吸附能随变化曲线Fig. 3 The CO adsorption energies for FeCu(CO)n (n=1-8)
分步吸附能和 FeCu 键长与 CO 数目的关系如图3 和图4所示. 从图中我们可以明显发现单分子吸附时的吸附能最大, 虽然三分子吸附比第二分子吸附能要大一些, 但整体趋势都是减小的;键长的变化则较为复杂, 两分子吸附时比一分子要长, 三分子和四分子时减小, 之后将逐步增加. 图 5给出了 FeCu(CO)n(n=0-8)最稳定结构的前线轨道能极差, 能极差越大, 表明把电子从最高占据轨道激发到最低空轨道越困难, 相应的结构也就越稳定. 从 HLG 曲线可以看出, 配合物 FeCO)4Cu(CO)3具有最大的 HLG 值, 结构最稳定. Mulliken电荷布居分析如图6所示, 其中红色圆点表示Fe原子的电荷密度, 黑色方点表示Cu原子的电荷密度, 从图中不难发现, FeCu吸附CO生成Fe(CO)4Cu(CO)1和继续吸附CO生成Fe(CO)4Cu(CO)3时Fe电荷密度均增大, 两者中电荷密度最大的是Fe(CO)4Cu(CO)1, 为2.125. Cu电荷密度在吸附过程中整体表现为增大, 直到生成Fe(CO)4Cu(CO)3, 此时Cu电荷密度为0.833.
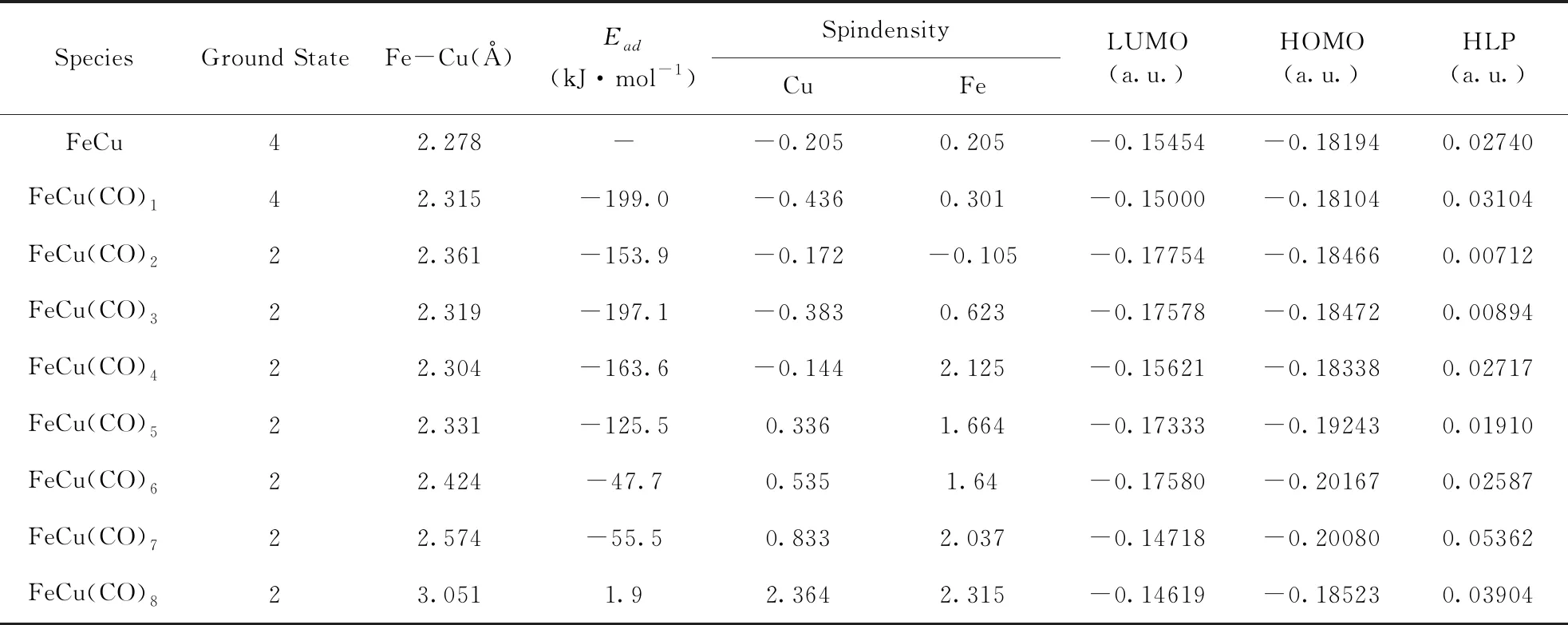
表2 FeCu(CO)n(n=0-8)各结构的基态多重度、FeCu键长、分步吸附能、Mulliken电荷分布, HOMO和LUMO的能量以及前线轨道能级差

图4 FeCu(CO)n (n=0-8)的Fe-Cu键长变化曲线Fig. 4 The binding energy as a function of FeCu(CO)n(n=0-8)
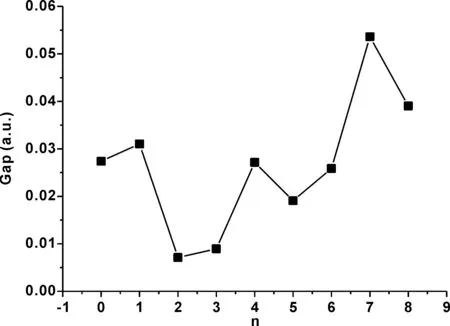
图 5 FeCu(CO)n (n=0-8)的前线轨道能级差Fig. 5 The HOMO-LUMO gaps of FeCu(CO)n (n=0-8)

图 6 FeCu(CO)n (n=0-8)的电荷密度Fig. 6 The Mulliken spin densities of FeCu(CO)n (n=0-8)
4 结 论
采用密度泛函理论的BPW91方法, 在6-311+G(d,p)基组水平上研究了FeCu团簇吸附CO过程中可能的几何结构和电子态. 结果表明: FeCu双原子团簇饱和吸附CO分子数为7, 其中生成FeCuCO的吸附能为-199.0 kJ·mol-1, 是该团簇反应过程最大吸附能. CO首先在Fe原子上吸附生成 (CO)4Fe-Cu, 吸附过程中金属原子满足18电子规则,对CO的吸附位置起主要决定作用. 配合物 Fe(CO)4Cu(CO)1和Fe(CO)4Cu(CO)3中Fe和Cu的电荷密度分别为2.125和 0.833, 在整个吸附过程中分别表现为最大.
猜你喜欢
数学物理学报(2022年3期)2022-05-25
数学物理学报(2022年1期)2022-03-16
陶瓷学报(2021年5期)2021-11-22
数学物理学报(2021年5期)2021-11-19
数学物理学报(2021年3期)2021-07-19
青岛大学学报(工程技术版)(2019年2期)2019-09-10
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16
中学化学(2015年8期)2015-12-29
应用化工(2014年1期)2014-08-16
生物加工过程(2013年1期)2013-03-11
