
密度泛函理论研究镉的二卤化合物分子的结构和振动频率
2016-01-16 03:05涂喆研
陕西理工大学学报(自然科学版) 2015年4期
关键词:键长
密度泛函理论研究镉的二卤化合物分子的结构和振动频率
涂喆研1,2
(1.西安工程大学 理学院, 陕西 西安 710048;
2.陕西省大分子科学重点实验室,陕西师范大学 化学化工学院, 陕西 西安 710062)
[摘要]利用12种泛函,结合大基组aug-ccpvqz(-pp),系统地计算研究了镉的二卤化合物分子CdX2(X=F,Cl,Br,I)的结构和振动频率,并与实验值进行对比。得到了相当高精度的关于CdX2分子的结构和振动频率的理论结果,并评估了这12种泛函用于计算这4个分子的结构和性质的适用性。研究结果显示:对于CdBr2分子,CAMB3LYP和PBE0泛函更适用于研究其结构和性质;对于CdI2分子,CAMB3LYP,M052X和PBE0泛函更适用于研究其结构和性质;对于CdF2分子,由于缺少键长的实验值,不下定论哪种泛函更适用于研究其结构和性质;对于CdCl2分子,一些泛函适用于计算其结构,而另一些泛函适用于计算其性质。但总体而言,CAMB3LYP和PBE0泛函的表现最为优秀。
[关键词]镉的二卤化合物;键长;振动频率;密度泛函理论;相对论效应
[文章编号]1673-2944(2015)04-0045-04
[中图分类号]O641
收稿日期:2015-04-03
基金项目:陕西省教育厅科学研究计划项目(2013JK0679);西安工程大学博士科研启动金资助项目(BS1211)
作者简介:涂喆研(1984—),男,江西省南昌市人,西安工程大学讲师,陕西师范大学在站博士后,主要研究方向为理论与计算化学。

对金属卤化物分子结构和性质的研究不仅属于基础研究范畴,而且在制灯工业[8]和半导体工业中有重要的实践应用[9]。高纯度难熔性纳米材料的燃烧合成技术的发展也需要更深入地理解金属卤化物体系的性质[10]。Hargittai[11]对金属卤化物的结构和性质的实验和理论研究作了综述,指出实验上用于确定结构的电子散射实验的成本太高,因此理论分析和预测金属卤化物的结构和性质显得十分重要。
Cd元素属于IIB族元素,它和卤族元素形成的二卤化物分子CdX2(X=F,Cl,Br,I)是稳定的离子化合物。CdX2的结构和振动频率的实验结果在文献[11]里已有报道。至于理论结果,最近的工作都要追溯到11年前。该课题组用Hartree-Fock(HF)方法和密度泛函(B3LYP)方法研究了CdX2分子的结构和振动频率[12]。但是,他们用的基组和赝势(ECPs)的精度不够,而且B3LYP泛函并不一定适用于计算所有的CdX2分子。因此,很显然他们的理论结果不能较好地吻合实验结果。近十年来,随着赝势、基组和密度泛函理论的发展,笔者认为选择高精度的赝势和与之匹配的大基组,并合理地使用泛函方法,会得到更加合理的理论结果。在本研究中,笔者用12种泛函研究CdX2分子的结构和振动频率。本研究工作有两个目的:第一,得到与实验值吻合得较好的CdX2分子的结构和振动频率的理论结果;第二,评估这12种泛函用于计算CdX2分子的结构和振动频率的适用性。
1计算细节
在本文的计算中,对Cd、Br和I原子使用赝势,而F和Cl原子没有使用赝势。标量相对论效应,也就是最内层电子的相对论效应用赝势参数描述。对Br原子使用ECP10MDF赝势[13],它的1s,2s和2p电子的相对论效应用赝势参数描述。对Cd[14]原子和I[15]原子使用ECP28MDF赝势,它们的1s,2s,2p,3s,3p和3d电子的相对论效应用赝势参数描述。对Cd[14]原子,Br[13]原子和I[15]原子使用与赝势相匹配的大基组aug-cc-pvqz-pp。对F[16]原子和Cl[17]原子使用全电子基组aug-cc-pvqz。
本文所采用的12种泛函分别为B3LYP,CAMB3LYP,X3LYP,M05,M052X,M06,M062X,PBE,PBE0,BPW91,BP86和LSDA。本文的理论计算均在Gaussian09程序包[18]中完成。
2结果和讨论
2.1结构

表1 理论计算的CdX 2分子的键长 (单位:Å)
CdX2分子是线性分子,具有D∞h对称性,已有其他课题组的理论计算证实了这一点[12]。理论计算的键长列在表1中,笔者采用较严格的误差标准,即理论值和实验值的误差不超过0.01 Å,那么并不是所有的泛函都得到了合理的键长。对CdF2分子,由于缺少实验值,笔者不下定论哪种泛函得到的键长更加合理。对CdCl2分子,泛函B3LYP,X3LYP,M05,M052X,M062X,BPW91和BP86计算得到的键长和实验值吻合得较好,误差小于0.01 Å。对CdBr2分子,泛函CAMB3LYP,M06和PBE0计算得到的键长和实验值吻合得较好,误差小于0.01 Å。对CdI2分子,泛函CAMB3LYP,M052X和PBE0计算得到的键长和实验值吻合得较好,误差小于0.01 Å。综上所述,泛函CAMB3LYP和PBE0将分子量最大的两个分子的键长均算得比较精准。泛函CAMB3LYP是长程校正版B3LYP,使用了库伦衰减方法。泛函PBE0使用了25%交换和75%关联的权重。
2.2振动频率
由于线性结构,CdX2分子有4个基本振动模式,其中2个用∑表示,分别为∑g和∑u,均为Cd—X键的拉伸振动。另外2个振动是简并的,记为∏u,是弯曲振动。振动频率的计算结果列在表2中,笔者同样采用较严格的误差标准,即理论值和实验值的误差不超过10 cm-1,那么并不是所有的泛函都能得到合理的振动频率。对CdF2分子,12种泛函均高估了振动频率∏u,其中,泛函M06得到了与实验值最接近的结果140 cm-1,较实验值121 cm-1相差19 cm-1。另一方面,泛函CAMB3LYP和PBE0得到了合理的振动频率∑g和∑u,与实验值的误差小于10 cm-1。对CdCl2分子,泛函CAMB3LYP,M06和PBE0得到了合理的振动频率,与实验值的误差均小于10 cm-1。对CdBr2分子,泛函CAMB3LYP,X3LYP,M05,M052X,M062X和PBE0得到了合理的振动频率,与实验值的误差均小于10 cm-1。对CdI2分子,泛函CAMB3LYP,M05,M052X,M06,M062X,PBE0和LSDA得到了合理的振动频率,与实验值的误差小于10 cm-1。综上所述,泛函CAMB3LYP和PBE0将4个分子的振动频率均算得比较精准。泛函CAMB3LYP是长程校正版B3LYP,使用了库伦衰减方法。泛函PBE0使用了25%交换和75%关联的权重。
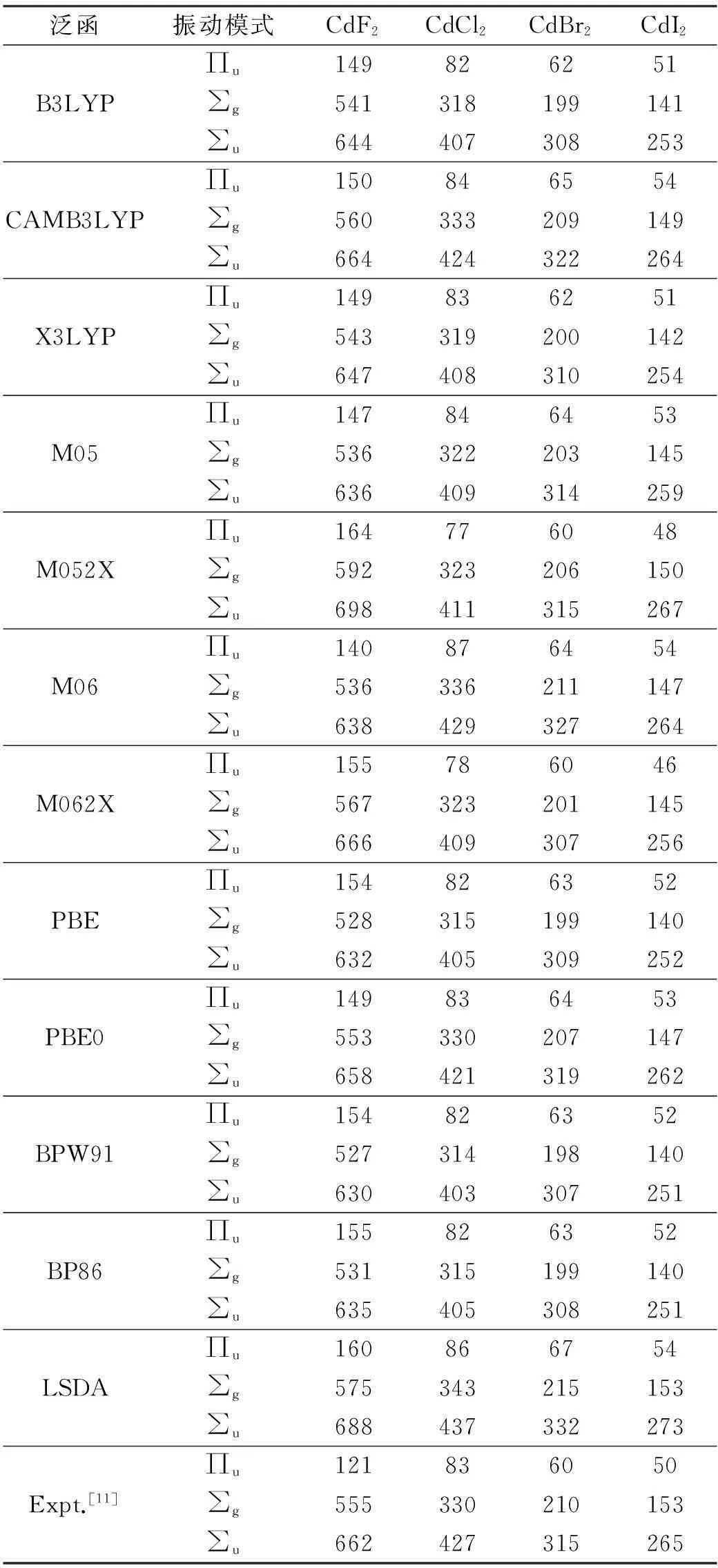
表2 理论计算的CdX 2分子的振动频率 (单位:cm -1)
3结论
在本文的工作中,笔者使用12种泛函来研究CdX2(X=F,Cl,Br,I)分子的结构和振动频率。笔者采用较严格的误差标准,即键长和振动频率的理论值与实验值的误差分别不超过0.01 Å和10 cm-1。从表1和表2中的数据以及之前的数据分析可以看出,泛函CAMB3LYP和PBE0最适合计算CdX2分子体系的结构和性质。
[参考文献]
[1]KULLIE O.A relativistic time-dependent density functional study of the excited states of the mercury dimer[J].Journal of Chemical Physics,2014,140(2):024304.
[2]SENN H M,BLÖCHL P E,Togni A.Toward an alkene hydroamination catalyst:static and dynamic ab initio DFT studies[J].Journal of the American Chemical Society,2000,122(17):4098-4107.

[4]SCHROEDER D,WESENDRUP R,HERTWIG R H,et al.Equilibrium isotope effects in cationic transition-metal(I) ethene complexes M(C2X4)+with M=Cu,Ag,Au and X=H,D[J].Organometallics,2000,19(13):2608-2615.
[5]BENCZE É,PáPAIA I,MINK J,et al.Spectroscopic and theoretical study of [PdCl3(C2H4)]-and [PdCl3(C2D4)]-complexes[J]. Journal of Organometallic Chemistry,1999,584(1):118-121.
[6]CITRA A,ANDREWS L.A spectroscopic and theoretical investigation of charge transfer complexes between silver and nitric oxide:infrared spectra and density functional calculations of AgNO+,0,-and Agx(NO)y Clusters (x,y=1,2) in Solid Argon and Neon[J].Journal of Physical Chemistry A,2001,105(13):3042-3051.
[8]HILPERT K,NIEMANN U.High temperature chemistry in metal halide lamps[J].Thermochimica Acta,1997,299(1/2):49-57.
[9]BENAVIDES-GARCIA M,BALASUBRAMANIAN K.Bond energies,ionization potentials,and the singlet-triplet energy separations of SnCl2,SnBr2,SnI2,PbCl2,PbBr2,PbI2,and their positive ions[J].Journal of Chemical Physics,1994,100(4):2821-2830.
[10]VORE De T C,GOLE J L.Energetics,molecular electronic structure,and spectroscopy of forming Group IIA dihalide complexes[J].Chemical Physics,1999,241(2):221-238.
[11]HARGITTAI M.Molecular structure of metal halides[J].Chemical Reviews,2000,100(6):2233-2302.
[12]ZHAO Jian-ying,ZHANG Yu,KAN Yu-he,et al.Theoretical studies on vibrational spectra of some halides of Group IIB elements[J].Spectrochimica Acta Part A:Molecular and Biomolecular Spectroscopy,2004,60(3):679-688.
[13]PETERSON K A,FIGGEN D,GOLL E,et al.Systematically convergent basis sets with relativistic pseudopotentials.II.Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16—18 elements[J].Journal of Chemical Physics,2003,119(21):11113-11123.
[14]PETERSON K A,PUZZARINI C.Systematically convergent basis sets for transition metals. II. Pseudopotential-based correlation consistent basis sets for the group 11 (Cu,Ag,Au) and 12 (Zn,Cd,Hg) elements[J].Theoretical Chemistry Accounts,2005,114(4/5):283-296.
[15]PETERSON K A,SHEPLER B C,FIGGEN D,et al.On the spectroscopic and thermochemical properties of ClO,BrO,IO,and their anions[J].Journal of Physical Chemistry A,2006,110(51):13877-13883.
[16]KENDALL R A,DUNNING Jr T H,HARRISON R J.Electron affinities of the first‐row atoms revisited. Systematic basis sets and wave functions[J].Journal of Chemical Physics,1992,96(9):6796-6806.
[17]WOON D E,DUNNING Jr T H.Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon[J].Journal of Chemical Physics,1993,98(2):1358-1371.
[18]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.Gaussian 09,Revision A.02[CP/CD]. Wallingford CT:Gaussian,Inc.,2009.
[责任编辑:谢 平]
Structure and vibration frequencies of cadmium dihalide molecules:
an evaluation of various density functional
TU Zhe-yan1,2
(1.School of Science, Xi’an Polytechnic University, Xi’an 710048, China
2.Key Laboratory for Macromolecular Science of Shaanxi Province,
School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi’an 710062, China)
Abstract:Compared with experimental results, 12 kinds of density functional based on aug-ccpvqz(-pp) basis set are used to study the structures and vibration frequencies of CdX2(X=F,Cl,Br,I) molecules systematically in this work. We provide the rather high-precision theoretical results of structures and vibration frequencies for CdX2 molecules. And we evaluate the qualification of different density functional in calculating the structures and properties of CdX2 molecules. It can be concluded that CAMB3LYP and PBE0 functional are more suitable for calculating CdBr2 molecule. CAMB3LYP, M052X, and PBE0 functional are recommended preferentially for calculating CdI2 molecule. Due to the absence of experimental bond length, it is difficult to determine which functional is more reliable for calculating both the structure and properties of CdF2 molecule. For CdCl2 molecule, some functionals are suitable for calculating its bond length while some other functionals are suitable for calculating its properties. But in one word, the performances of CAMB3LYP and PBE0 functional are the best.
Key words:cadmium dihalide;bond length;vibration frequency;density functional theory;relativistic effect
猜你喜欢
四川大学学报(自然科学版)(2022年4期)2022-07-22
山东化工(2020年8期)2020-06-12
青岛大学学报(工程技术版)(2019年2期)2019-09-10
原子与分子物理学报(2019年1期)2019-03-19
山东化工(2018年20期)2018-11-08
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
化工技术与开发(2015年8期)2015-10-25
火炸药学报(2012年4期)2012-01-29
合成化学(2010年2期)2010-11-26
