
CuCl催化苯腙和丙炔酸甲酯Michael加成反应的机理研究
2019-03-19 09:21刘柳斜王海峰李来才
原子与分子物理学报 2019年1期
陈 晓, 刘柳斜, 王海峰, 郑 妍, 李来才
(四川师范大学化学与材料科学学院,成都 610066)
1 引 言
Michael加成反应是有机化学中非常重要高效的C-C和C-杂原子构建反应[1,2]. 该反应是合成药物,材料中间体及化工产品的重要反应,因此Michael 加成反应一直受到化学研究者的关注[3,4]. 吡唑环类化合物是一类重要的医药、农药、材料合成中间体, 一直以来备受制药与合成工业青睐[5-8].吡唑类化合物的社会需求巨大,而如何快速、高效、经济 、环保的合成吡唑类化合物是相关研究的热点之一. Fricero等人研究了吡唑杂环化合物的合成中间体5-三氟化硼吡唑盐的合成和系列杂环合成反应[9]. 最近,李玉峰等人通过肼、醛和丙炔酸甲酯三组分串联反应建立了一种高效的构建吡唑类杂环的方法,其中由丙炔酸甲酯对亚胺进行Michael加成而形成的中间体产物是设计串联式反应构建氮杂环的关键中间体[10]. 通过在氯化亚铜催化下,丙炔酸甲酯和亚胺进行Michael加成的方法形成吡唑合成中间体. 该反应具有简单,高效,原料价廉易得的特点.由于对于苯腙和丙炔酸甲酯发生Michael 加成反应的机理研究还未见报道,为了解该反应的本质,本文采用密度泛函理论探讨了苯腙和丙炔酸甲酯进行Michael 加成的微观反应机理. 由于理论计算方法一直以来都是对化学反应微观机理研究的有利工具, 我们课题组曾通过理论计算方法分别研究过Pt10团簇催化肉桂醛选择加氢反应和Rh的芳基化合物与含氮芳基卤代物交叉偶联反应的相关机理,均得到了可信赖的结果[11,12]. 同样本文希望通过理论计算方法对今后相关的学术研究提供可信的理论基础.
2 计算原理和方法
本文采用密度泛函理论[13-15](DFT)中的M05方法,在6-31+G(d) ( Cu采用赝势基组 LanL2DZ)基组水平上,分析了反应过程中的能量变化,探讨了苯腙和丙炔酸甲酯发生Michael加成反应的微观反应历程. 反应进程中的所有化合物均进行了全参数优化和频率计算,[16,17]以确定每个过渡态均有唯一虚频. 通过频率振动分析确认了中间体和过渡态的合理性, 内禀反应坐标 (instrinsic reaction coordinate, IRC) 跟踪计算对各个过渡态连接的对应的反应物和产物进行了确认; 通过自然键轨道(NBO)[18]理论分析了分子的轨道间相互作用, 为反应的进一步进行提供了理论数据. 理论计算压强和温度设置均为程序默认的1 atm和298.15 K. 理论计算均采用 Gaussian 09[19]程序包完成.
3 结果分析
本研究根据反应可能发生的位点设计了两条反应通道I1和I2,分别对应苯腙的N原子和C原子. 总共涉及4个过渡态,为确认相关过渡态的真实性,通过振动分析得到了各自唯一的虚频率. 两条反应通道优化的反应物,中间体和过渡态的构型见图1.
3.1 反应机理分析
苯腙与丙炔酸甲酯反应活性位点主要在苯腙分子上的碳活性位点和氮活性位点. 因此在氯化亚铜催化下的苯腙与丙炔酸甲酯发生Michael加成反应存在着两种可能的路径. 但这两种反应方式的起始物都是M1,中间体M1是由氯化铜和反应物R1和R2络合形成的. 中间体M1中Cu-N(1), Cu-C(2), Cu-C(3)的键长分别为0.2134 nm, 0.1989 nm, 0.2032 nm. 中间体M1中N(1)-N(2) 键长比反应物R1增加了0.0015 nm, C(2)-C(3)键长比R2增加了0.0034 nm.轨道理论(NBO)分析可知在中间体M1中, BD(2)C(2)-C(3)→LP*(6)Cu二阶稳定化能E(2)为41.10 kcal/mol, LP(1)N(1)→LP*(5)Cu二阶稳定化能E(2)为131.96 kcal/mol, 表明它们之间存在轨道间的相互作用. 由表1中能量数据可以看出, 中间体M1的能量分别比R1和R2及催化剂CuCl的能量之和低45.32 kcal/mol,中间体M1易形成并能够稳定存在, 完成了催化活化过程.在反应通道I1中由M1→TS1→M2, 中间体M1通过过渡态TS1形成中间体M2, 活化能为34.86 kcal/mol,是反应的速控步骤. 在TS1中N(2)-H(2)和C(3)-H(2)的键长分别为0.1337 nm 和0.1276 nm. 表明化学键N(2)-H(2)正在断裂而化学键 C(3)-H(2)正在形成, 即H(2)正在从N(2)向C(3)转移.而C(2)-C(3)键长为0.1284 nm 比中间体M1的C(2)-C(3)增加了0.0038 nm,表明原来的炔键正在向烯键转变.在中间体M2中和Cu-C(2)的键长是0.1933 nm, 比过渡态TS1减少了0.0033 nm, 而Cu-N(2) 键长为0.1953 nm,表明Cu-N(2)正在形成. 而C(3)-C(2)的键长为0.1344 nm比TS1中的键长增加了0.0060 nm 此时碳碳双键已经形成. 在接下来的M2→TS2→M3反应过程中, 从M2到TS2, Cu-N(2)键长变长了0.0033 nm, Cu-C(2) 键长变短了0.0013 nm, C(2)-N(2)的键长是0.2267 nm. 这表示了C(2)-N(2)键正在形成.在中间体M3中C(2)-N(2)的键长是0.1351 nm, C(2)-C(3)的键长是0.1397 nm.它们与过渡态TS2相比分别减少了0.0916 nm和增加了0.0063 nm.
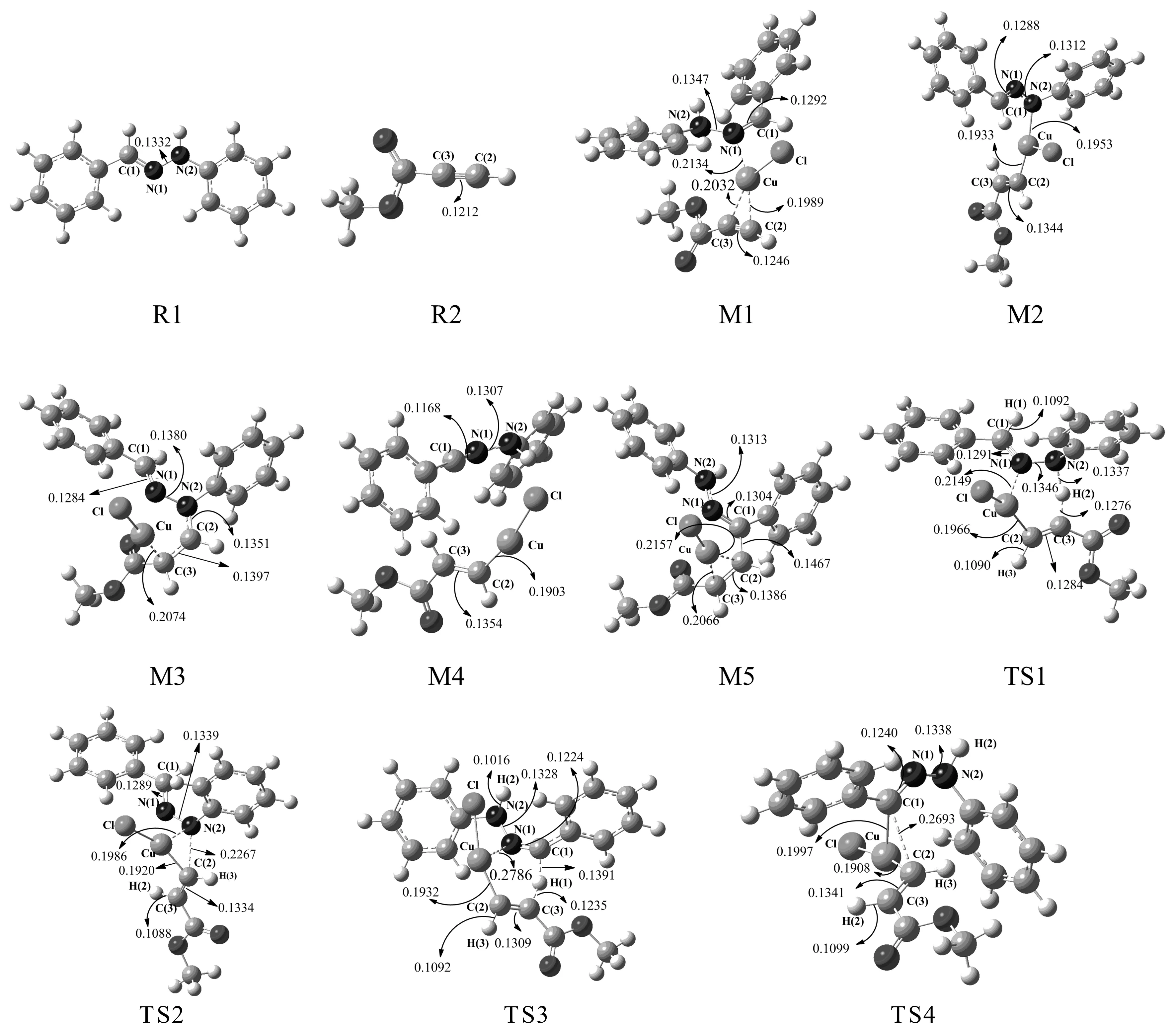
图1 反应路径中各驻点的分子构型图(单位为nm)Fig. 1 Geometric parameters of the compounds (nm)
这意味着在中间体M3中C(2)-N(2)已经形成,而原来的碳碳双键向碳碳单键变化,相应的Michael加成产物已经形成. 在反应通道I2中由M1→TS3→M4,在过渡态TS3中Cu-N(1) 和Cu-C(2)的键长分别为0.2786 nm和0.1932 nm. Cu-N(1) 键长比中间体M1中增加了0.0652 nm, Cu-C(2)的键长相比于M1而言减少了0.0057 nm.可以看出Cu-N(1) 键在逐渐断裂. TS3中的C(1)-H(1)和H(1)-C(3)键长分别是0.1391 nm和0.1235 nm.表明H(1)正在从C(1)向C(3)迁移.而且TS3中的C(2)-C(3)键长也变长了0.0063 nm, M1中的炔键正在向烯键转变. TS3中的N(1)-C(1)键长是0.1224 nm,比M1变短了0.0068 nm, 这预示着原来的碳氮双键正在向三键变化. 在中间体M4中C(2)-C(3)键长比TS3增加了0.0045 nm,此时碳碳双键已经形成. M4中的Cu-N(1)键已经断裂,N(1)-C(1)键长为0.1168 nm,比TS3减少了0.0056 nm, 表明碳氮三键已经形成. 在M4→TS4→M5变化过程中,TS4中C(1)-N(1)键长是0.1240 nm, 比M4增加了0.0072 nm, 预示着碳氮三键正在向碳氮双键变化,C(1)-C(2)和Cu-C(1)的键长分别是0.2693 nm和0.1908 nm,表示C(1)-C(2)和Cu-C(1)正在成键. 在中间体M5中C(1)-N(1)键长是0.1304 nm,比过渡态TS4增加了0.0064 nm, 碳氮双键已经形成. C(1)-C(2)键长是0.1467 nm比TS4减少了0.1226 nm, C(1)-C(2)已经成键. C(2)-C(3)键长是0.1386 nm增加了0.0045 nm,碳碳双键向碳碳单键变化对应的Michael加成产物已经形成.
3.2 反应过程能量分析
反应路径中各驻点能量E (a.u.),相对能量Erel(kcal/mol) 及频率v (cm-1)见表1. 由表1中能量数据可以看出, 反应物(R1,R2)与催化剂CuCl结合形成中间体M1的过程, 能量降低了45.32 kcal/mol, 表明中间体M1能稳定存在.在反应通道I1中中间体M1通过过渡态TS1形成中间体M2的过程中活化能是34.86 kcal/mol,是反应的速控步骤. 中间体M2通过过渡态TS2形成中间体M3的过程中活化能是6.17 kcal/mol. 在反应通道I2中中间体M1通过过渡态TS3形成中间体M4的过程中活化能是41.94 kcal/mol,是反应的速控步骤.中间体M4通过过渡态TS4形成中间体M5的过程中活化能是-1.13 kcal/mol. 通过对比两条反应路径的活化能,表明反应通道I1的活化能较低反应更容易进行.
表1 反应各驻点的能量E(a.u.),相对能量Erel(kcal/mol) 及频率v(cm-1)
Table 1 EnergiesE(a.u.), relative energiesErel(kcal/mol) and frequenciesv(cm-1) of the compounds in the reactions

PathSpeciesEErelvR1+R2+CuCl-1570.7255940—M1-1570.797811-45.32—TS1-1570.742266-10.46-1300.51I1M2-1570.757519-20.03—TS2-1570.747674-13.86-162.63M3-1570.847733-76.64—TS3-1570.730976-3.38-261.13I2M4-1570.763841-24.00—TS4-1570.763981-25.13-51.28M5-1570.851274-78.87—
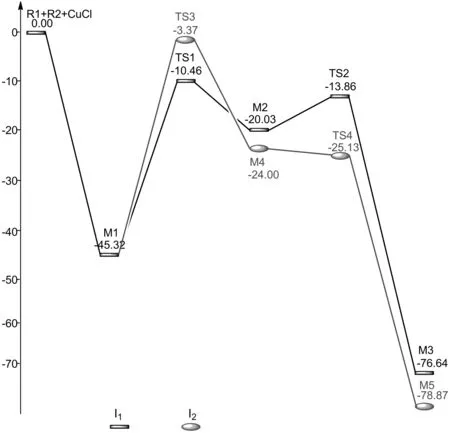
图2 反应能级图(单位为kcal/mol)Fig. 2 The schematic of energy levels (kcal/mol)
4 结 论
本文采用密度泛函理论中的M05方法对苯腙和丙炔酸甲酯在CuCl催化下发生Michael加成反应机理进行了研究. 针对不同的反应位点,我们设计了两组反应路径I1和I2. 经过对这两组反应路径中由各个驻点相对能量分析,苯腙和丙炔酸甲酯在CuCl催化下,反应通道I1控制步骤是M1→TS1→M2,活化能为34.86 kcal/mol, 反应通道I2控制步骤是M1→TS3→M4,活化能为41.94 kcal/mol, 反应通道I1所需活化能更低, 反应能够顺利进行. 我们的研究结果从理论上解释了苯腙和丙炔酸甲酯在CuCl催化下发生Michael加成反应特征,解释了实验现象.
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
油气·石油与天然气科学(2021年12期)2021-12-11
合成树脂及塑料(2020年4期)2020-09-20
洛阳理工学院学报(自然科学版)(2020年1期)2020-05-15
青岛大学学报(工程技术版)(2019年2期)2019-09-10
电脑知识与技术(2018年3期)2018-03-21
分析化学(2017年12期)2017-12-25
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
枣庄学院学报(2015年5期)2016-01-09
