
藏橐吾药材质量标准研究
2021-12-30 03:14万玉莹周燕达娃卓玛西藏大学拉萨8500中国科学院成都生物研究所成都6004中药藏药质量控制重点实验室西藏自治区食品药品检验研究院拉萨850005
中南药学 2021年11期
万玉莹,周燕,达娃卓玛,3*(.西藏大学,拉萨 8500;.中国科学院成都生物研究所,成都 6004;3.中药藏药质量控制重点实验室 西藏自治区食品药品检验研究院,拉萨 850005)
藏医药学是我国民族医药学的瑰宝,藏药质量标准问题一直是制约藏药发展的核心问题,其中藏药材的标准化更是藏药现代化的前提和基础[1-2]。藏橐吾[Ligularia rumicifolia(Drumm.)S.W.Liu]是菊科(Compositae)橐吾属(Ligularia)多年生草本,主要分布在我国的西藏东部及尼泊尔海拔3700~4500 m的灌丛、山坡等地方[3],全草入药[4],是藏药体系的常用药材,资源丰富[5-6]。西藏珞巴族将其作为消炎药治疗咽喉疼痛[7],但目前对其报道相对较少。藏橐吾性温,味苦,入肺经,常与其他藏族当地的橐吾属植物一起被称作“日肖”,用于清热解毒、缓解烧伤及风湿等症状[4],其药用价值在《中国中药资源志要》中也有所记载,温肺止咳,润肺降气,还有散寒、化痰以及止痰的功效[5]。
历版《中国药典》、各地方药典、《卫生部药品标准·藏药·第一册》正文中均未收载藏橐吾相关质量标准。本研究根据《中国药典》2020年版第四部通则相关规定[8],首次对藏橐吾药材根、茎、叶及粉末的显微特征和薄层色谱进行鉴别,并对药典规定的常规项目进行检查,确定使用藏橐吾药材中大量且具有生物活性的倍半萜成分——蜂斗菜素为薄层鉴别及含量测定指标[9],为地方药材质量标准的制订提供参考,为国家药材质量标准进一步完善奠定基础。
1 材料
1.1 仪器与试药
Waters Acquity UPLC H-CLASS液相色谱仪(四元泵,PDA检测,柱温箱,自动进样器,Empower 色谱数据工作站);色谱柱为Welch Ultimate XBC18(250 mm×4.6 mm,5 μm);Sartorius BT 25 S(十万分之一)、BSA124S-CW(万分之一)电子天平(德国赛多利斯公司);蜂斗菜素对照品(实验室自制,纯度>99.0%);硅胶G板(青岛海洋化工有限公司);甲醇、乙腈为色谱纯(中国MORELLK公司);水为超纯水;其余试剂均为分析纯。
1.2 药材
藏橐吾药材8批次,均采自西藏自治区境内各不同地区,经西藏自治区藏医院格桑巴珠副主任药师鉴定为菊科橐吾属植物藏橐吾[Ligularia rumicifolia(Drumm.)S.W.Liu],药材全草晒干,粉碎后过2号筛,混合均匀,备用。样品信息见表1。
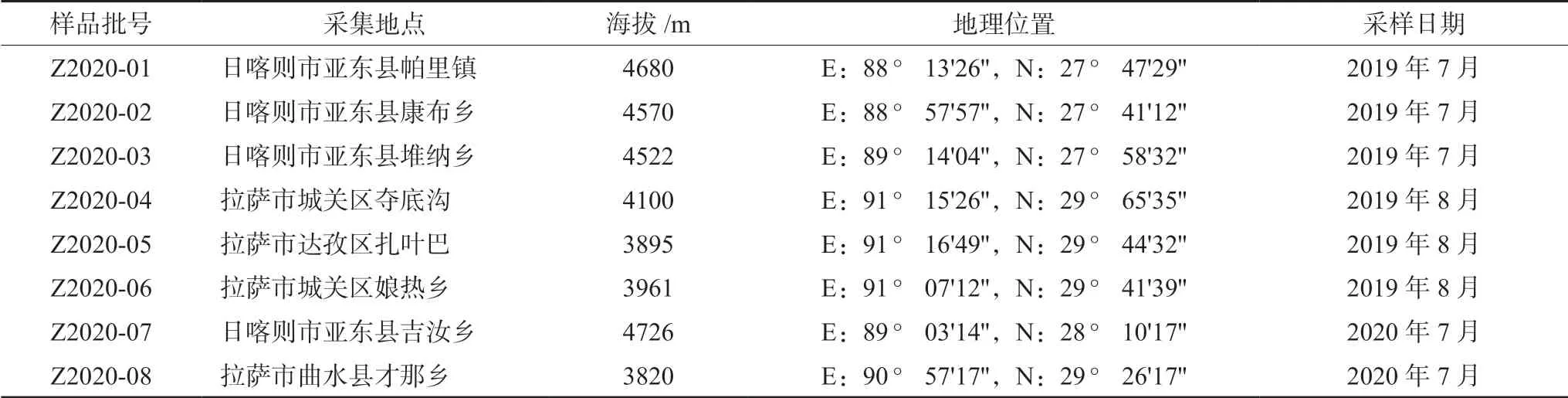
表1 藏橐吾药材采集信息 Tab 1 Sample collection information of Ligularia rumicifolia (Drumm.) S.W.Liu
2 方法与结果
2.1 药材性状特征
药材干燥根部呈不规则块状,大小不一,根肉质,多数,根簇生多数细根,长3~15 cm,直径0.1~0.3 cm,表面灰褐色,有纵皱纹,质较柔韧,不易折断。叶片椭圆形或卵形,长15~20 cm,宽5~7 cm,叶基呈心形,边缘具规则的三角形齿;茎生叶无柄,基部抱茎,表面棕褐色,近圆柱形,质地较硬,易折断,内部淡黄色,中空结构。气微香,味苦。藏橐吾原植物及干燥药材见图1。

图1 藏橐吾原植物(A)及干燥药材(B)Fig 1 Original plant(A)and dry medicinal material(B)of Ligularia rumicifolia(Drumm.)S.W.Liu
2.2 显微特征
2.2.1 根横切面 呈类圆形,表皮细胞1~2列,直径约24 μm;外周孔隙较大,直径为0.05~0.1 mm,环绕一周有5~7个;皮层多为薄壁细胞,直径约48 μm,15~17层疏松排列;内周孔隙呈类圆形,周状排列一周5~7个,直径0.05~0.1 mm;中间有中心维管束,直径约为横切面直径的1/6。韧皮部呈圆环形排列在木质部周围,5~7列;木质部呈紧密排列圆形,约占横切面半径的1/16(见图2)。
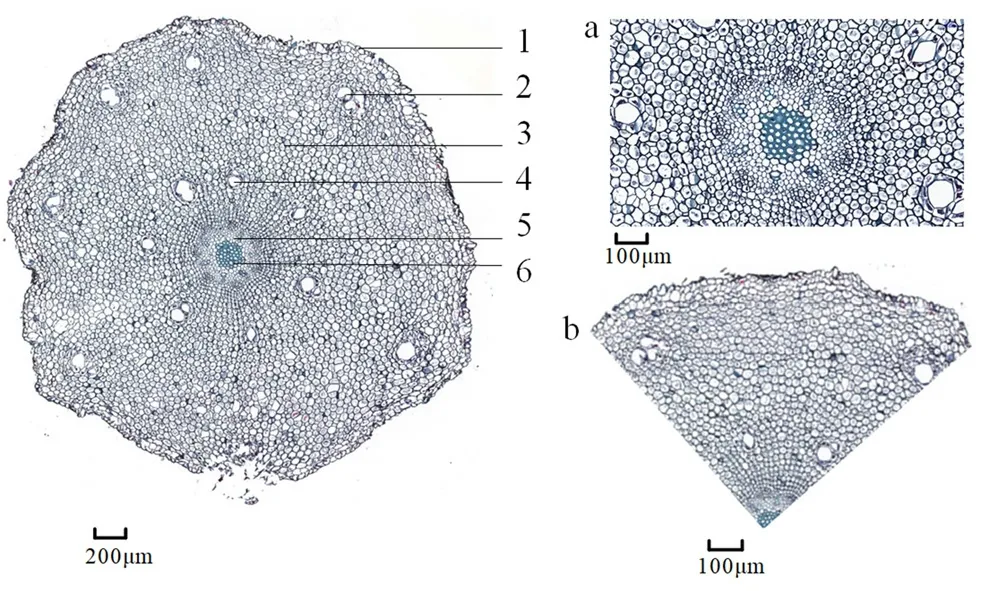
图2 藏橐吾根横切面Fig 2 Cross section of Ligularia rumicifolia(Drumm.)S.W.Liu root
2.2.2 茎横切面 类圆形,表皮细胞1列,皮层较厚,4~5列排列成环;双韧性维管束周状排列,一周环绕15~20个,2个旁支上各有3~4个双韧性维管束;髓部由数十列细胞组成的环髓带包围。中心类圆形导管,半径约为横切面半径的1/10(见图3)。

图3 藏橐吾茎横切面Fig 3 Cross section of Ligularia rumicifolia(Drumm.)S.W.Liu stem
2.2.3 主叶脉横切面 上、下表皮细胞各1列,椭圆形或类圆形;海绵组织细胞3~5列,不规则疏松排列;栅栏组织紧密排列2~3列;中间有中心维管束,椭圆形,木质部呈疏松排列半圆形,直径约0.2 mm,细胞大小不一;韧皮部在木质部下方,韧皮部细胞紧密排4~5列,细胞较木质部细胞较小,直径约为木质部细胞1/2(见图4)。

图4 藏橐吾主叶脉横切面Fig 4 Main vein cross section of Ligularia rumicifolia(Drumm.)S.W.Liu
2.2.4 生药粉末 本品粉末灰褐色或灰绿色,叶表皮细胞分两种,一种是上表皮,细胞呈方形或多角形,另一种是下表皮,细胞呈波浪形,气孔不定式,副卫细胞4~5个,大小30.0~61.6 μm。纤维单个或成束存在,木纤维具纹孔。薄壁细胞类圆形,大小不等。木栓细胞类圆形,壁增厚。非腺毛较多,表面光滑。螺纹导管为主,也存在网纹及具缘纹孔导管,直径29.8~42.9 μm。木栓石细胞多见,表面观呈类长方形、长条形或多边形,垂周壁连珠状增厚,长110.1~143.5 μm,宽15.4~33.9 μm,可见细密的裂纹及纹孔。散在草酸钙方晶,直径35.3~55.4 μm。棕色块不规则状,大小不等,散在(见图5)。

图5 藏橐吾生药粉末Fig 5 Crude drug powder of Ligularia rumicifolia(Drumm.)S.W.Liu
2.3 薄层鉴别
称取藏橐吾药材粉末1.0 g,加入25 mL 甲醇,常温超声45 min,放置室温,过滤,取续滤液作为供试品溶液。另称取蜂斗菜素适量,加甲醇溶解制成质量浓度为0.68 mg·mL-1的对照品溶液。照薄层色谱法(通则0502),吸取上述溶液各 1 μL,点于同一硅胶 G 薄层板,以石油醚(60~90℃)-乙酸乙酯(3∶1)为展开剂,展开,取出,晾干,喷25%磷钼酸溶液,105℃加热至斑点显色清晰。可在样品、对照药材及对照品相应位置上,观察到相同深蓝色斑点(见图6)。
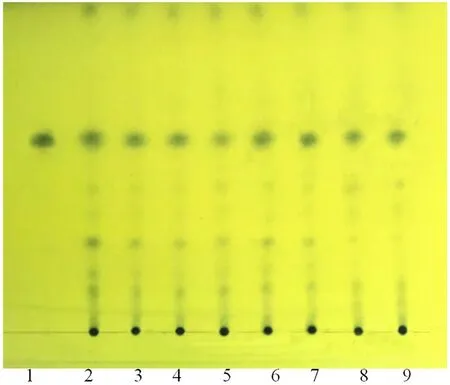
图6 藏橐吾TLC图Fig 6 TLC of Ligularia rumicifolia(Drumm.)S.W.Liu
2.4 水分测定
不同产地的藏橐吾药材水分含量8.58%~ 10.12%,平均值含量为9.4%。以平均值上浮20%为上限,暂定本品水分不得过 11.0%。8批样品水分含量均在规定范围内,药材水分合格率为100%(通则0832 第二法)。结果见表2。
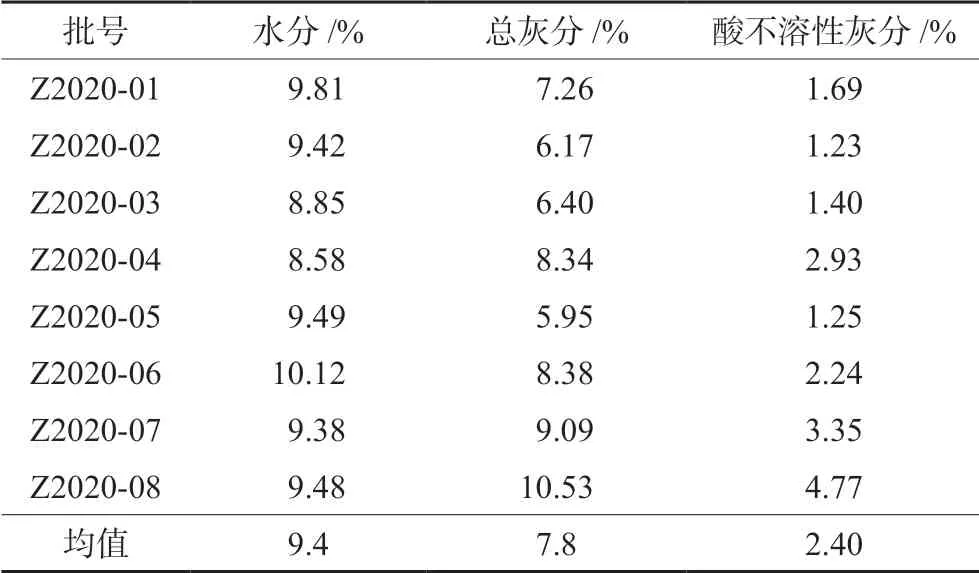
表2 水分、总灰分、酸不溶性灰分的测定结果(n=2) Tab 2 Determination of water,total ash and acid insoluble ash (n=2)
2.5 灰分、酸不溶性灰分测定
不同产地的藏橐吾药材总灰分含量为5.95%~10.53%,平均值为7.8%,酸不溶性灰分含量为1.23%~4.77%,平均值为2.40%。以平均值上浮20%为上限,暂定本品总灰分含量不得过 9.5%,酸不溶性灰分含量不得过3.0%。其中在8批藏橐吾药材中,药材批号为Z2020-08总灰分不合格,药材批号为Z2020-08、Z2020-07酸不溶性灰分不合格。药材总灰分合格率为87.5%;酸不溶性灰分合格率为75%(通则2302),结果见表2。
2.6 浸出物测定
随机选取一批药材为代表(批号:Z2020-03),对浸出物提取方法进行考察。结果表明,以浸膏计,热浸法以纯水作为溶剂时得到浸出物含量最高。采用此方法测得8批不同产地藏橐吾药材水溶性浸出物含量为 44.30%~50.96%,平均值为45.3%,以本品干燥品平均值的80%为下限,暂定本品水溶性浸出物不得少于32.3%。8批不同产地的藏橐吾药材均符合要求(通则2201)。药材浸出物合格率为100%,结果见表3~4。
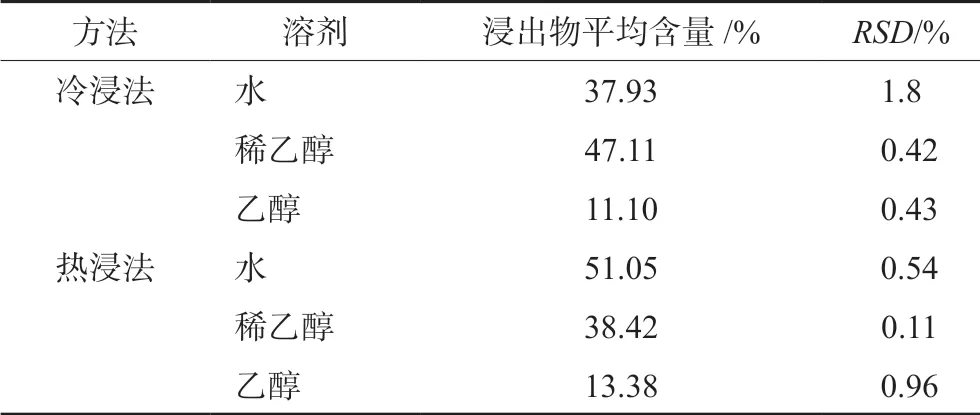
表3 浸出物提取方法的考察(n=2) Tab 3 Extraction method (n=2)
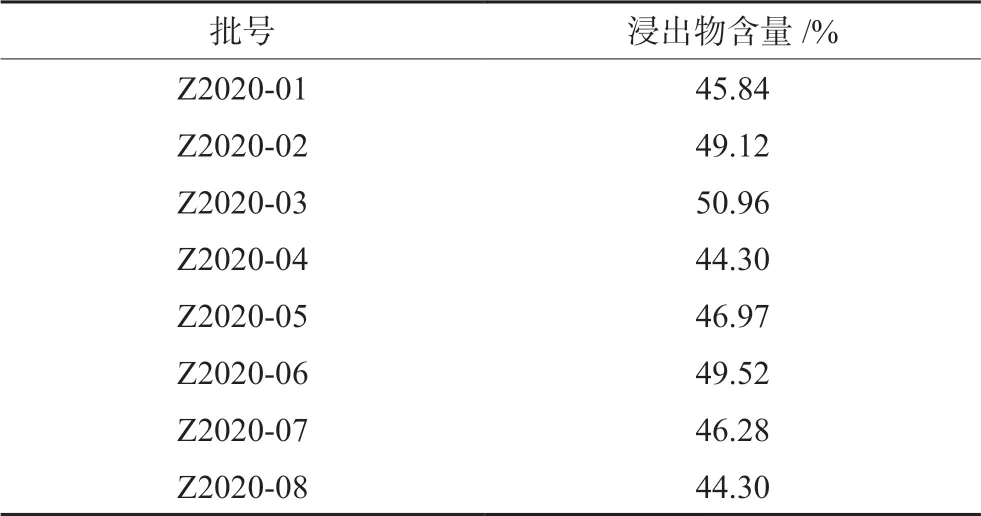
表4 水溶性浸出物含量测定结果(n=2) Tab 4 Determination of water-soluble extract content (n=2)
2.7 蜂斗菜素含量测定
2.7.1 色谱条件 流动相:甲醇-乙腈-0.1%甲酸 水=38∶43∶19(V/V/V);色 谱 柱:Welch Ultimate®XB-C18(250 mm×4.6 mm,5 μm);流速:1.0 mL·min-1;检测波长:232 nm;柱温:25℃;进样量:10 μL。
2.7.2 对照品溶液的制备 精密称取蜂斗菜素对照品适量,用甲醇稀释得到质量浓度为815.6 μg·mL-1的对照品储备液。精密吸取对照品储备液1 mL用甲醇定容到25 mL量瓶中,得到质量浓度为32.62 μg·mL-1的对照品溶液。
2.7.3 供试品溶液的制备 精密称取藏橐吾药材粉末(过2号筛)0.5 g,精密加入甲醇25 mL,称量。常温超声45 min,放置室温,称量,用甲醇补足重量;过滤,续滤液用0.22 μm微孔滤膜过滤,作为供试品溶液。
2.7.4 系统适用性试验 精密吸取对照品溶液、供试品溶液、空白溶液(甲醇)进样测定。结果显示,蜂斗菜素与相邻色谱峰的分离度均>1.5,与其他组分可以达到完全分离,对称因子在0.95~1.05,峰形对称,理论塔板数≥10 000。结果见图7。

图7 蜂斗菜素HPLC色谱图Fig 7 HPLC chromatogram of petasin
2.7.5 线性关系考察 精密量取对照品储备液,制成7个蜂斗菜素对照品系列溶液,以对照品质量浓度(X,μg·mL-1)为横坐标,峰面积Y为纵坐标,进行线性回归,得到回归方程为Y=7.853×104X+8.241×104(r=0.9996),结果表明,蜂斗菜素在9.79~45.67 μg·mL-1内与峰面积线性关系良好。
2.7.6 精密度考察 取对照品溶液连续进样6次,记录峰面积。结果蜂斗菜素峰面积的RSD为0.60%,表明该仪器精密度良好。
2.7.7 重复性考察 选择同一批次药材(批号:Z2020-06),按“2.7.3”项下方法制备供试品溶液平行试验6次,结果显示蜂斗菜素含量的RSD为0.63%,表明该方法重复性良好。
2.7.8 稳定性考察 取“2.7.7”项下供试品溶液于0、2、6、8、10、12 h 进样,记录峰面积。结果蜂斗菜素的峰面积RSD为0.47%,表明供试品溶液在12 h内稳定。
2.7.9 加样回收试验 精密称取已知含量的藏橐吾(批号:Z2020-06)样品9份,每份约 0.25 g,按供试品中待测成分含量的80%、100%、120%精密加入蜂斗菜素对照品,进样测定,结果蜂斗菜素加样回收率分别为96.41%、96.26%、95.47%,RSD分别为1.5%、1.5%、1.8%,表明方法准确度良好。
2.7.10 样品测定 分别取西藏不同产地的藏橐吾药材,按“2.7.3” 项下方法制备供试品溶液,进样测定,外标法计算藏橐吾药材中蜂斗菜素的含量。结果8批药材中蜂斗菜素含量范围 0.116%~0.168%,平均含量为0.141%。以平均含量的80%为限(按干燥品计算),暂定藏橐吾样品中蜂斗菜素含量不得少于0.1%。8批不同产地的藏橐吾药材均符合要求,结果见表5。

表5 蜂斗菜素含量测定结果(n=3) Tab 5 Content determination of petasin (n=3)
3 讨论
3.1 对照品的选择
据文献报道,藏橐吾主要化学成分有黄酮类[10]、单萜[10]、生物碱类[11]、倍半萜类[9,12-15]等,且认为从橐吾属植物中分离出的倍半萜类物质是抗菌的主要活性成分[13]。
Ye等[9]从藏橐吾药材中分离出45个倍半萜类成分,艾里莫酚烷型的倍半萜蜂斗菜素作为其中一个大量且特征性成分,具有抑菌、抗炎[16]、舒张血管[17]、抗肿瘤[18-21]、解痉、抗白三烯、抗组胺[22]等多种药理活性。因此,考虑临床功效、含量、特征性和分离难度等因素,本课题组将蜂斗菜素作为指标成分,进行藏橐吾药材质量标准研究。
3.2 薄层色谱鉴别
3.2.1 展开剂、薄层板类型的考察 考察了5种不同类型展开剂系统:石油醚(60~90℃)-乙酸乙酯(3∶1)、石油醚(60~90℃)-丙酮(5∶1)、石油醚-二氯甲烷-乙酸乙酯(2∶0.5∶0.5)、二氯甲烷-乙酸乙酯(4∶1)、二氯甲烷-甲醇(10∶1),结果在石油醚(60~90℃)-乙酸乙酯(3∶1)和石油醚(60~90℃)-丙酮(5∶1)展开剂体系下,分离效果较好,且Rf值适中,相比之下,前者分离效果和对照物质成点性更好,Rf值0.65左右,展开后斑点清晰明显,无拖尾,符合药典相关规定。
3.2.2 显色剂的考察 考察4种显色剂显色效果(25%磷钼酸溶液、5%硫酸-乙醇溶液、2% FeCl3-乙醇溶液、1%香兰素-硫酸溶液)。结果25%磷钼酸溶液作显色剂展开后斑点清晰,背景无干扰。
3.3 含量测定
3.3.1 供试品溶液制备方法的考察 采用不同浓度的甲醇、乙醇溶液(50%、70%、100%)作为提取溶剂,当提取溶液是100%甲醇时,得到蜂斗菜素含量最高。在此基础上,考察冷浸、超声、回流三种提取方式在不同提取时间下对蜂斗菜素提取含量的影响,结果表明超声45 min时,提取效率最高。
3.3.2 流动相的考察 考察了甲醇-水、甲醇-甲酸水(0.1%、0.2%)、乙腈-水、乙腈-甲酸水(0.1%、0.2%)等二元流动相体系,发现很难将目标峰与周围峰很好分离,在此基础上考察三元流动相体系甲醇-乙腈-水、甲醇-乙腈-甲酸水(0.1%、0.2%),结果综合考虑压力、峰形以及样品分离效果等因素,最终确定流动相为甲醇-乙腈-0.1%甲酸水=38∶43∶19。
3.4 小结
本研究通过对多产地藏橐吾药材性状描述、显微鉴别及初次建立薄层鉴别方法,可知该植物鉴别特征明显,可作为藏橐吾药材的鉴别依据;通过对藏橐吾药材各检查项及浸出物测定,初步确定各常规试验项限量;通过首次建立蜂斗菜素对照品的含量测定方法,初步确定藏橐吾药材质控指标性化学成分及限量,为其品种鉴别、地方药材质量标准的制订及进一步开发利用提供依据。
猜你喜欢
煤化工(2022年3期)2022-07-08
选煤技术(2022年2期)2022-06-06
选煤技术(2022年2期)2022-06-06
广州化工(2022年1期)2022-01-26
选煤技术(2021年6期)2021-04-19
四川蚕业(2021年4期)2021-03-08
广州化工(2020年23期)2020-12-16
贵州科学(2020年5期)2020-09-02
中国资源综合利用(2016年10期)2016-01-22
大家健康(学术版)(2012年2期)2012-10-10
