
一种结构新颖的中氮茚衍生物的合成新方法
2022-01-21 06:19叶青青
安徽师范大学学报(自然科学版) 2021年6期
叶青青
(滁州城市职业学院 医学系,安徽 滁州 239000)
引言
中氮茚是一类非常重要的含氮杂环骨架,广泛存在于天然产物和活性药物分子中[1-2]。许多中氮茚衍生物具有重要的生物或药物活性,如抗炎性[3]、抗癌活性[4]、抗菌及抗氧化性[5]、抗肿瘤活性等[6]。近年来,一些中氮茚衍生物由于在可见光区域具有长波长吸收性能和高的荧光量子产率被发现在分子探针领域具有广泛应用[7]。因此,发展条件温和、步骤经济性和操作简单的构建中氮茚骨架的方法引起了有机化学家和药物化学家的研究兴趣。
传统方法中,构建中氮茚骨架有两种途径:(1)利用吡啶衍生物构建吡咯环,有N-亚甲基吡啶叶立德与缺电子烯烃或炔烃的1,3-偶极环加成反应[8-12],金属催化吡啶衍生物的分子内环异构化反应等[13-15];(2)利用吡咯衍生物构建吡啶环[16-18]。目前,使用1,3-偶极环加成反应是构建中氮茚骨架最重要的方法。但是这些方法往往受底物多样性、需要使用金属催化剂等实验条件的限制。多组分反应具有步骤和原子经济性,可通过一步反应构建多个化学键而成为复杂活性杂环化合物合成的一种有效途径[19-21]。本文以价廉易得的吡啶作为碱和反应底物,基于其与溴代苯乙酮和叠氮基肉桂酸酯类化合物的三组分串联反应,实现一种结构新颖的1,2,3-三取代中氮茚衍生物的高效合成,设计合成路线如图1所示。
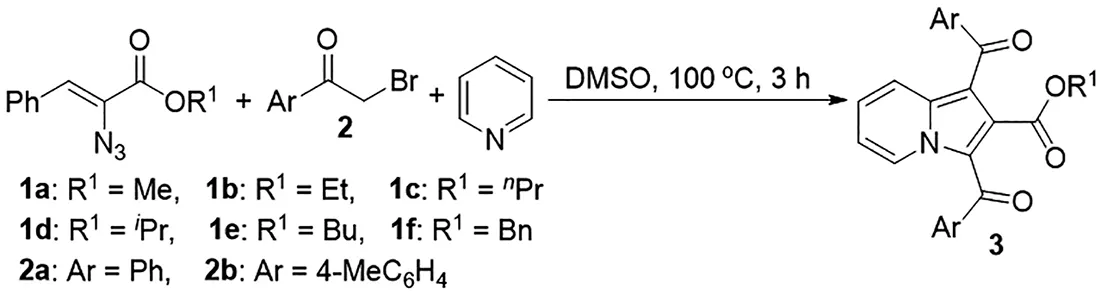
图1 1,2,3-三取代中氮茚衍生物的设计合成
1 实验部分
1.1 仪器与试剂
核磁共振仪(BRUKER-AV-300型,CDCl3为溶剂,TMS为内标);显微熔点测定仪(X-4型,北京泰克仪器有限公司,温度计未经校正)。
实验所用试剂和溶剂均为市售化学纯或分析纯(可直接使用)。
1.2 1,2,3-三取代中氮茚类化合物3a~3k的合成
取易干燥的Schlenk管,向其中加入叠氮肉桂酸酯(0.5mmol),溴代苯乙酮(1.0mmol),吡啶(0.75mmol)和二甲亚砜(DMSO,3mL),在搅拌下将反应管放入预先加热至100℃的油浴中反应3小时,TLC跟踪反应,反应结束后,将反应体系冷却至室温,加入10mL水,并用二氯甲烷萃取(3×10mL),合并有机相,无水Na2SO4干燥2~3小时,过滤后溶液真空浓缩,剩余物通过硅胶柱层析[洗脱剂:石油醚/乙酸乙酯=8/1至3/1(v/v)]进行分离,可以得到目标化合物1,2,3-三取代中氮茚衍生物3a~3k。
1,3-二苯甲酰基中氮茚-2-甲酸甲酯(3a):黄色固体,产率71%;m.p.132~133℃;1H NMR(300MHz,CDCl3)δ:9.63(d,J=7.1Hz,1H),8.22(d,J=9.0Hz,1H),7.72(d,J=7.4Hz,3H),7.58~7.49(m,3H),7.42~7.40(m,5H),7.16(t,J=7.0Hz,1H),2.82(s,3H);13C NMR(75MHz,CDCl3)δ:191.1,187.1,164.5,151.1,140.2,140.0,138.0,132.1,132.0,128.5,128.2,127.7,127.6,119.9,116.3,51.7;HR-MS(ESI-TOF)m/z:Calcd for C24H18NO4{[M+H]+} 384.1236,found 384.1240。
1,3-二苯甲酰基中氮茚-2-甲酸乙酯(3b):黄色固体,产率78%;m.p.129~130℃;1H NMR(300MHz,CDCl3)δ:9.55(d,J=6.3Hz,1H),8.16(d,J=8.6Hz,1H),7.70(d,J=6.5Hz,3H),7.48~7.39(m,8H),7.12(t,J=4.8Hz,1H),3.12(q,J=6.9Hz,2H),0.73(t,J=6.9Hz,3H);13C NMR(75MHz,CDCl3)δ:191.1,187.1,164.1,151.0,140.2,140.0,138.0,133.6,132.1,130.1,129.1,128.6,128.4,128.2,127.6,127.4,119.8,116.2,61.4,13.1;HR-MS(ESI-TOF)m/z:Calcd for C25H20NO4{[M+H]+} 398.1392,found 398.1395。
1,3-二苯甲酰基中氮茚-2-甲酸正丙酯(3c)黄色固体,产率79%;m.p.123~124℃;1H NMR(300MHz,CDCl3)δ:9.56(d,J=7.1Hz,1H),8.19(d,J=9.0Hz,1H),7.70(d,J=7.2Hz,3H),7.52~7.44(m,3H),7.41~7.35(m,5H),7.11(t,J=7.0Hz,1H),2.95(t,J=6.7Hz,2H),1.19~1.08(m,2H),0.69(t,J=7.4Hz,3H);13C NMR(75MHz,CDCl3)δ:191.1,187.1,164.2,140.1,140.0,138.0,132.1,130.2,128.7,128.2,127.6,127.5,121.0,119.8,116.2,113.3,67.1,21.0,10.3;HR-MS(ESI-TOF)m/z:Calcd for C26H22NO4{[M+H]+} 412.1549,found 412.1546。
1,3-二苯甲酰基中氮茚-2-甲酸异丙酯(3d):黄色固体,产率72%;m.p.130~131℃;1H NMR(300MHz,CDCl3)δ:9.40(d,J=6.9Hz,1H),8.00(d,J=9.0Hz,1H),7.76(d,J=7.1Hz,3H),7.58~7.30(m,8H),7.04(t,J=6.6Hz,1H),4.13~4.05(m,1H),0.63(d,J=6.0Hz,6H);13C NMR(75MHz,CDCl3)δ:191.0,187.2,163.7,140.1,139.7,132.4,130.1,129.1,129.0,128.4,127.3,126.9,119.6,115.9,70.1,20.7;HR-MS(ESI-TOF)m/z:Calcd for C26H22NO4{[M+H]+} 412.1549,found 412.1548。
1,3-二苯甲酰基中氮茚-2-甲酸正丁酯(3e):黄色固体,产率61%;m.p.135~136℃;1H NMR(300MHz,CDCl3)δ:9.55(d,J=6.7Hz,1H),8,18~8.10(m,2H),7.70(d,J=6.9Hz,3H),7.50~7.48(m,3H),7.40~7.38(m,4H),7.10(t,J=6.4Hz,1H),3.01(d,J=6.3Hz,2H),1.10~0.98(m,4H),0.78(t,J=6.9Hz,3H);13C NMR(75MHz,CDCl3)δ:191.2,187.2,164.3,140.1,139.9,138.0,133.6,132.2,132.1,130.1,128.7,128.5,128.3,128.2,127.6,119.8,116.3,65.5,29.7,19.0,13.6;HR-MS(ESI-TOF)m/z:Calcd for C27H24NO4{[M+H]+} 426.1705,found 426.1706。
1,3-二苯甲酰基中氮茚-2-甲酸苯甲酯(3f):黄色固体,产率54%;m.p.122~123℃;1H NMR(300MHz,CDCl3)δ:9.55(d,J=6.8Hz,1H),8.19(d,J=8.7Hz,1H),7.69(d,J=6.7Hz,3H),7.51~7.41(m,4H),7.35~7.26(m,7H),7.11(t,J=6.5Hz,1H),6.97(s,2H),4.00(s,2H);13C NMR(75MHz,CDCl3)δ:191.1,187.1,164.0,140.1,139.9,138.0,134.3,133.6,132.2,132.1,130.1,128.7,128.4,128.3,128.2,127.6,119.8,116.3,67.3;HR-MS(ESI-TOF)m/z:Calcd for C30H22NO4{[M+H]+} 460.1549,found 460.1544。
1,3-二(4-甲基苯甲酰基)中氮茚-2-甲酸甲酯(3g):黄色固体,产率75%;m.p.128~129℃;1H NMR(300MHz,CDCl3)δ:9.53(d,J=7.0Hz,1H),8.13(d,J=9.0Hz,1H),7.64~7.61(m,4H),7.40(t,J=7.6Hz,1H),7.25~7.21(m,4H),7.11(t,J=6.9Hz,1H),2.89(s,3H),2.41(s,6H);13C NMR(75MHz,CDCl3)δ:190.8,186.9,164.6,142.9,142.7,137.7,137.5,137.3,128.9,128.8,128.7,127.6,127.1,119.8,116.0,51.7,21.5;HR-MS(ESI-TOF)m/z:Calcd for C26H22NO4{[M+H]+} 412.1549,found 412.1553。
1,3-二(4-甲基苯甲酰基)中氮茚-2-甲酸乙酯(3h):黄色固体,产率71%;m.p.120~121℃;1H NMR(300MHz,CDCl3)δ:9.46(d,J=7.0Hz,1H),8.09~7.98(m,1H),7.85~7.78(m,1H),7.62(d,J=7.5Hz,1H),7.37~7.18(m,7H),7.05(t,J=6.6Hz,1H),3.21(q,J=6.9Hz,2H),2.36(s,3H),0.75(t,6.9Hz,3H);13C NMR(75MHz,CDCl3)δ:190.9,186.9,164.3,150.9,143.0,142.8,137.7,137.6,137.3,130.1,129.1,128.9,127.5,127.0,119.7,115.9,61.4,21.6,13.2;HR-MS(ESI-TOF)m/z:Calcd for C27H24NO4{[M+H]+} 426.1705,found 426.1702。
1,3-二(4-甲基苯甲酰基)中氮茚-2-甲酸正丙酯(3i):黄色固体,产率74%;m.p.127~128℃;1H NMR(300MHz,CDCl3)δ:9.47(d,J=6.5Hz,1H),8.10(d,J=8.6Hz,1H),7.62(d,J=7.0Hz,4H),7.35(t,J=7.0Hz,1H),7.26~7.19(m,4H),7.05(t,J=6.4Hz,1H),3.06(t,J=6.0Hz,2H),2.37(s,6H),1.19~1.12(m,2H),0.69(t,J=6.7Hz,3H);13C NMR(75MHz,CDCl3)δ:190.8,186.9,164.4,142.9,142.8,137.7,137.5,137.3,128.9,127.5,126.9,119.8,115.9,67.1,21.5,21.0,10.4;HR-MS(ESI-TOF)m/z:Calcd for C28H26NO4{[M+H]+} 440.1862,found 440.1865。
1,3-二(4-甲基苯甲酰基)中氮茚-2-甲酸异丙酯(3j):黄色固体,产率64%;m.p.125~126℃;1H NMR(300MHz,CDCl3)δ:9.32(d,J=6.9Hz,1H),7.94(d,J=8.9Hz,1H),7.67(d,J=7.1Hz,3H),7.30~7.19(m,6H),7.00(t,J=6.5Hz,1H),4.22~4.13(m,1H),2.37(s,6H),0.67(d,J=6.1Hz,6H);13C NMR(75MHz,CDCl3)δ:190.8,187.0,163.8,143.3,142.2,137.4,137.2,137.1,130.2,129.3,129.2,129.1,129.0,127.2,126.4,119.5,115.6,69.9,21.6,20.7;HR-MS(ESI-TOF)m/z:Calcd for C28H26NO4{[M+H]+} 440.1862,found 440.1867。
1,3-二(4-甲基苯甲酰基)中氮茚-2-甲酸正丁酯(3k):黄色固体,产率68%;m.p.123~124℃;1H NMR(300MHz,CDCl3)δ:9.46(d,J=6.7Hz,1H),8.11(d,J=8.7Hz,1H),7.62(d,J=7.3Hz,4H),7.35(t,J=7.3Hz,1H),7.26~7.17(m,4H),7.05(t,J=6.4Hz,1H),3.10(t,J=6.9Hz,2H),2.36(s,6H),1.10~1.08(m,4H),0.78(t,J=6.9Hz,3H);13C NMR(75MHz,CDCl3)δ:190.9,186.9,164.4,142.9,142.8,137.7,135.5,137.3,130.2,129.8,129.1,128.9,127.5,127.0,119.8,115.9,65.4,29.7,21.6,19.0,13.6;HR-MS(ESI-TOF)m/z:Calcd for C29H28NO4{[M+H]+} 454.2018,found 454.2013。
2 结果与讨论
首先,以叠氮基肉桂酸乙酯(1b,1 equiv.),溴代苯乙酮(2a,2 equiv.)和吡啶(1 equiv.)为模板底物对实验条件进行了优化,具体结果见表1。首先,在二甲亚砜为溶剂,100℃下反应3小时,仅能以54%的收率获得目标产物(表1,序号1)。随后在反应体系中增加吡啶的用量至1.2倍当量时,产率升高至65%(表1,序号2),继续增加吡啶的用量至1.5倍当量时,产率达到最高78%(表1,序号3)。由此表明,吡啶在反应体系中不仅作为反应物参与反应,也作为碱促进反应的进行。然后,考察了其他碱对该反应的影响,使用1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU),1,4-二氮杂二环[2.2.2]辛烷(DABCO)和4-二甲氨基吡啶(DMAP)等有机碱时,反应产率均较低(表1,序号5~7)。

表1 三组分串联反应合成1,2,3-三取代中氮茚化合物反应条件的优化
使用NaHCO3和Na2CO3作碱时,反应很难进行,仅能检测到痕量的产物生成(表1,序号8和9)。进一步地研究发现,该反应仅能在二甲亚砜溶剂中较好的进行,在其他溶剂中,如甲苯(toluene),N,N-二甲基甲酰胺(DMF),1,4-二氧六环(1,4-dioxane),乙醇和1,2-二氯乙烷(1,2-DCE),该反应很难进行(表1,序号10~14)。最后,在对反应温度和反应时间的优化中发现,该反应在100℃下反应3小时为最好(表1,序号15~21),产率达到最高78%。
随后在最优化实验条件下,对底物的普适性进行了探究,具体见图2。分析目标产物结构发现,在反应过程中叠氮基肉桂酸酯中苯环并未进入产物结构中,因此,重点考察了叠氮基肉桂酸酯中酯基对该反应的影响。实验结果表明,各种烷基叠氮基肉桂酸酯类化合物均能很好的发生反应(如—Me,—Et,—nPr,—iPr,—nBu),并以61%~79%的产率得到目标产物3a~3e,即使使用具有较大位阻的叠氮基肉桂酸异丙酯作为底物,也能以72%的产率获得目标产物。使用叠氮基肉桂酸苯甲酯作为底物时,由于苄基位阻的影响,仅能以54%的产率获得目标产物3f。随后使用对甲基溴代苯乙酮作为底物,在标准实验条件下也能以64%~76%的产率得到相应的1,2,3-三取代中氮茚衍生物3g~3k。进一步考察了吡啶上取代基的影响,遗憾的是,无论吡啶4-位上连有给电子的甲基或吸电子的氯基团时,该反应均不能发生。

图2 底物拓展
根据底物结构、反应结果及已有文献报道[22-24],对该反应提出了一种可能的反应机理(图3)。首先,溴代苯乙酮2a与吡啶反应生成中间体A,同时,叠氮基肉桂酸乙酯1b在热驱动下释放氮气得到中间体B,继而发生中间体A对中间体B的亲核加成/开环反应得到中间体C。接着,中间体C发生消除反应得到中间体D。中间体A在碱作用下生成中间体E,并与中间体D发生1,3-偶极环加成反应得到中间体F,随后中间体F在吡啶的作用下脱去质子得到中间体G,中间体G进一步发生分子内消除反应(利用GC-MS在反应体系中捕捉到苄胺的存在)得到中间体H,最后中间体H发生氧化芳构化反应生成最终产物3b。

图3 可能的反应机理
3 结论
发展了一种有机碱促进下,吡啶与叠氮基肉桂酸酯和溴代苯乙酮的三组分串联反应合成1,2,3-三取代中氮茚衍生物的新方法。反应过程中吡啶首先与溴代苯乙酮反应生成硫叶立德作为亲核试剂和1,3-偶极子前体,随后经1,3-偶极环加成反应/消除/芳构化历程得到中氮茚杂环骨架。该方法底物价廉易得,适用范围广,实验操作简单,为1,2,3-三取代中氮茚衍生物的合成提供了一种新策略。
猜你喜欢
中国药学药品知识仓库(2022年10期)2022-05-29
科学导报(2022年21期)2022-04-10
现代农药(2022年1期)2022-02-15
教育周报·教育论坛(2020年3期)2020-10-21
科技资讯(2018年16期)2018-10-26
科技信息·下旬刊(2018年8期)2018-10-21
山东工业技术(2018年9期)2018-05-26
科技资讯(2017年12期)2017-06-09
成长·读写月刊(2017年3期)2017-04-08
科技视界(2016年26期)2016-12-17
