
影响鸡脂肪肝出血综合征的因素及“多重打击”学说
2022-08-26 09:00涂闻君
畜牧兽医学报 2022年8期
张 弥,涂闻君,张 奇,江 莎,2*
(1. 西南大学动物医学院 动物健康与动物性食品安全国际合作联合实验室,重庆400715;2.西南大学 医学研究院免疫学研究中心,重庆402460)
鸡FLHS是鸡肝和腹腔内异位脂肪沉积过多,伴随肝组织质脆和出血为特征的营养代谢性疾病,病鸡在临床上表现为突然发病、肝破裂、出血和突然死亡等特征。剖检发现,约40%蛋鸡死亡由FLHS造成,而在笼养蛋鸡中,这个数据更高达74%,且97%FLHS病鸡有大量脂肪堆积。因此,该病是笼养鸡非感染性死亡的主要原因之一,给禽蛋业造成了严重的经济损失。鸡的肝组织在脂肪合成和代谢中起主要作用,肝组织中脂肪合成量远大于脂肪组织,经选育的商品鸡饲料转化率高,更易发生代谢紊乱。鸡FLHS的发生原因、机理、病理症状与哺乳动物非酒精性脂肪肝病(nonalcoholic fatty liver disease,NAFLD)类似,因此也常作为NAFLD研究的动物模型。本文从遗传、营养、激素、环境、肠道微生物等方面阐述可能引起鸡FLHS发生的原因,列举和对比研究人员通过不同方法等构建的FLHS模型。尽管鸡FLHS的病理机制尚不完全清楚,但可与NAFLD类似,如胰岛素抵抗、肝氧化应激和炎症反应等,被学者广泛接受的NAFLD发病机制“多重打击”学说可为解释FLHS提供一定的参考,本文从FLHS的脂肪变性、胰岛素抵抗、脂肪毒性、氧化应激、炎症、纤维化、脂肪因子分泌异常和自噬抑制等多重方面综述FLHS可能的发病机制,为进一步深入研究鸡FLHS提供参考。
1 影响鸡FLHS发生的因素
1.1 遗传与表观遗传
不同品系的鸡患FLHS的敏感程度存在差异。在蛋鸡中,Abplanalp和Napolitano报道白来航蛋鸡品系由于其肝组织中酶活性高,相比于同龄其他品系蛋鸡,容易发生肝功能障碍而诱发FLHS;肉鸡中,Zhang等报道京兴黄鸡比北京油鸡更易形成脂肪肝,且脂肪肝可从父系遗传给后代:第一代(F0)高脂饮食诱导FLHS,后代饲喂正常日粮,从F1代到F3代,表现出肝糖脂代谢关键基因上调,FLHS发病率是对照组的两倍。
另一方面,表观遗传变化与遗传风险因素相互作用决定疾病的易感性,肝特定DNA异常甲基化、miRNA和lncRNA都与脂质代谢和脂肪肝密切相关,为单纯性脂肪变性到NAFLD过程中的重要决定因素之一,但很少有关于其在FLHS进程中的报道。FLHS鸡的肝低甲基化特征已被确定,Liu等报道乙酰辅酶A羧化酶(acetyl CoA carboxylase,ACC)启动子的甲基化水平降低,可增加脂肪从头合成(lipogenesis,DNL),增加肝脂肪含量;微粒体三酰甘油转移蛋白(microsomal triglyceride transfer protein,MTTP)启动子的甲基化水平降低,促进MTTP表达而增加肝细胞的甘油三酯(triglyceride,TG)输出。Li等发现,代谢相关的lncRNA(lncLTR)由雌激素调节且与母鸡的血浆TG显著相关。Tan等研究显示,在高能日粮诱导FLHS鸡的肝组织中,检测观察的83种与脂代谢和糖代谢相关的基因中有72%上调,而检测观察的150种免疫相关基因中有81%下调,并发现23个靶基因(如1、3和)是甲基化和lncRNAs调控的中心基因。同样,Zhu等报道高能低蛋白日粮影响327(一种启动子的表观遗传标记)的组蛋白修饰和染色质结构,导致鸡肝脂质代谢、免疫应答和过氧化物酶体增殖物激活受体(peroxisome proliferators-activated receptors,PPAR)信号通路基因失调,使肝组织脂肪过度积累并诱发FLHS。然而还尚未有研究报道与肝纤维化的存在和程度相关的表观修饰变化。这些结果强调了表观遗传修饰对基因表达的调控,并在FLHS发挥核心调节作用。因此,鉴定关键基因和表观遗传标记可扩大对FLHS分子机制的研究。
1.2 营养
FLHS病因与营养密切相关,日粮中能量过高,饱和脂肪酸过多,缺乏氨基酸、矿物质和维生素等是诱发FLHS常见因素。
能量过高是导致FLHS 发生的重要原因之一,因商品鸡快速生长或高产蛋量需求,往往配比高能饲料;鸡淋巴系统不够完善,从肠道吸收的脂肪酸通过肝门静脉系统直接转运至肝,并且鸡肝组织中脂肪合成速度远胜于哺乳动物,超过90%的脂肪在肝内合成,因此,日粮中能量过高常诱导鸡发生FLHS。为提高鸡饲料的适口性和能量密度,饲料中的脂肪来源往往不同,而这也会影响FLHS的发生,不同脂肪酸对鸡FLHS发生具有不同的影响。脂肪酸主要包括饱和脂肪酸(saturated fatty acid,SFA)、单不饱和脂肪酸(monounsaturated fatty acid,MUFA)和多不饱和脂肪酸(polyunsaturated fatty acid,PUFA)。黄油、猪油等动物油是典型的SFA;油酸属于MUFA,橄榄油、牛油果油和茶油中含有大量的油酸。PUFA分为n-3 PUFA和n-6 PUFA,α-亚麻酸(ALA)、二十碳五烯酸(EPA)和二十二碳六烯酸(DHA)属于n-3 PUFA,亚麻籽油中含有丰富的ALA,鱼油中含有丰富的DHA;亚油酸(LA)属于n-6 PUFA,大豆油、玉米油、葵花籽油、菜籽油等植物油含有丰富的亚油酸。饲料中添加MUFA和PUFA可减少氧化应激和炎症表达、降低低密度脂蛋白胆固醇的浓度、提高胰岛素敏感性,而添加SFA可增加肝脂肪含量,诱导胰岛素抵抗。在代谢中n-6和n-3 PUFA互相竞争辅酶,饮食中大量n-6 PUFA代谢产生的花生四烯酸为炎症介质,相反,n-3 PUFA 中的EPA和DHA则促进免疫细胞产生特异性促炎症消退介质进而减少炎症,并提高肉鸡NK细胞活力和促进淋巴细胞增殖,n-6/n-3 PUFA比例的增加与肥胖和NAFLD有关,在蛋鸡日粮中添加n-3 PUFA则可丰富肠道微生物群和促进脂质代谢。FLHS进程中,三种不饱和脂肪酸(油酸、亚油酸、棕榈油酸)含量降低,其显著减少导致过氧化和氧化应激的增强,而过氧化和氧化应激增强可导致载脂蛋白B(apolipoprotein B,Apo B)的水解,进而损害极低密度脂蛋白(very low density lipoprotein,VLDL)的分泌,导致脂肪肝并减少卵黄中脂质营养物的沉积,阻碍卵黄的发育和排卵。进一步研究表明,亚油酸缺乏会引起生殖障碍疾病和脂质代谢紊乱,Fu等报道母体补充共轭亚油酸可减少鸡胚肝中甾醇调节元件结合蛋白-1c(sterol-regulatory element binding protein-1c,SREBP-1c)的蛋白表达,通过激活腺苷酸激活蛋白激酶(adenosine 5′-monophosphate (AMP)-activated protein kinase,AMPK)和PPARα信号通路降低鸡胚肝的脂质合成。因此,FLHS鸡的肝脂肪沉积和产蛋率下降可能与血清不饱和脂肪酸缺乏有关。
肝DNL背后的酶促反应和载脂蛋白的形成需要维生素和辅助因子的参与,如脂溶性维生素、胆碱、甲硫氨酸、磷脂、硒等。维生素E和膳食抗氧化混合剂可适度改善高氧化剂(3%氧化油、3%PUFA)日粮肉鸡肝功能损伤和炎症。胆碱、磷脂甜菜碱和叶酸作为甲基供体,饮食中缺乏这些物质可影响表观遗传修饰的稳定性,增加鸡FLHS的易感性;而日粮中补充胆碱则减少蛋鸡肝总脂质和TG含量,提高抗氧化能力;母体补充甜菜碱使雄性幼鸡免受皮质醇诱导的肝TG积累。甲硫氨酸和胆碱为磷脂酰胆碱的重要前体,磷脂酰胆碱是构成VLDL颗粒外壳的主要磷脂,甲硫氨酸/胆碱缺乏(methionine-choline deficiency,MCD)使VLDL合成受损、导致TG在肝细胞积累;因此,MCD也常用于诱导鸡FLHS模型。磷脂是组成生物膜的主要成分,其不同营养制品广泛应用于鸡饲料,饮食中添加3%大豆磷脂能有效改善FLHS病鸡相关肝和血液指标的异常变化。硒是禽类必需的微量元素,为谷胱甘肽过氧化物酶(glutathione peroxidase,GSH-Px)的重要成分,缺乏可导致鸡肝肿大、质脆和营养不良,Ren等研究表明,硒可提高GSH-Px活性,缓解砷诱导的肉鸡肝脂肪变性和肝损伤。
氨基酸通过三羧酸循环提供能量,并形成脂类转运所需的蛋白质,如Apo A和Apo B等载脂蛋白的形成需要多种氨基酸,与鸡FLHS密切相关;Guo等研究发现,鸡FLHS表现为血清丝氨酸含量增加和异亮氨酸、苏氨酸含量减少的氨基酸代谢紊乱,Zhuang等报道FLHS鸡肝组织丙氨酸、谷氨酰胺和苯丙酮酸等非必需氨基酸增加,而缬氨酸、异亮氨酸、苏氨酸和甲硫氨酸等必需氨基酸减少,表明机体可能通过吸收氨基酸促进脂类转运,而缺乏相关氨基酸导致鸡FLHS。
日粮中钙、磷和维生素D不足,可导致采食量增加、机体能量蓄积,并通过下丘脑抑制促性腺激素分泌,降低产蛋量,同时进一步导致肝脂肪的积累;及时补充钙、磷或使用营养补充剂提高钙、磷利用率等方法可缓解相关负面影响,并且补充维生素D还可增强鸡抗氧化活性和抗炎作用。
1.3 环境
笼养限制了鸡的活动,有数据显示,在笼养蛋鸡中,高达74%死亡鸡因FLHS引起,而自由放养模式中仅5%左右。春、夏季有更高的FLHS发病率,可能与环境温度有关。热应激通过抑制电子传递链活性,增加电子泄露使肝中活性氧(reactive oxygen species,ROS)水平升高,进而诱导氧化应激破坏肝脂质代谢。Lu等报道慢性热应激会增加肉鸡肝TG和总胆固醇(total cholesterol,TC)含量,上调脂肪酸合成相关基因如、脂肪酸合酶(fatty acid synthase,)、碳水化合物反应元件结合蛋白(carbohydrate response element binding protein,)和基因表达,激活肝核受体α(liver X receptor alpha,LXRα)途径使肝脂肪合成增加。
1.4 激素
17β-雌二醇(E)是主要的循环雌激素,在蛋鸡受光刺激后1~2 d增加,并在产蛋过程中保持较高浓度。E可促进肝合成大量游离脂肪酸(free fatty acid,FFA)、TG和胆固醇酯,组装成VLDL和卵黄原蛋白颗粒,通过血液循环输送到卵巢产生蛋黄前体。添加外源E可显著增加鸡肝中TG、TC含量,同时上调脂肪酸和胆固醇合成相关基因,肌肉注射E可诱导鸡FLHS模型。
慢性应激或长期暴露于高糖皮质激素(glucocorticoid,GC)可导致鸡脂肪肝,日粮中添加皮质醇(cortisol,CORT)使肉鸡血清葡萄糖、TG、TC和肝的相对重量、糖原含量增加,皮下注射CORT则通过激活脂肪合成、抑制线粒体脂肪酸β氧化导致鸡脂肪肝,并且长期CORT暴露可进一步引发鸡肝组织炎症和纤维化;同样,口服地塞米松(dexamethasone,DEX)抑制肉鸡三羧酸循环和脂肪酸氧化、促进脂肪酸从头合成导致糖脂代谢紊乱;注射DEX影响肉鸡肝组织AMPK和胆汁酸合成信号通路。
骨钙素(osteocalcin,OCN)由成骨细胞分泌,以非羧化形式(ucOCN)进入血液调节能量代谢。OCN可通过多条途径影响肝的脂质代谢,即促进脂联素和胰岛素分泌、提高胰岛素敏感性、激活核因子E2相关因子2(nuclear factor E2-related factor 2,Nrf2)途径、抑制c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)或核因子κB(Nuclear factor kappa-B,NF-κB)途径。Xia等研究显示,人类血清ucOCN水平与肝脂肪变性、炎症和纤维化分级呈负相关,外源性补充ucOCN则能够改善饲喂西方饮食小鼠的肝脂肪变性、降低SREBP-1及其下游的蛋白表达,表明OCN水平与NAFLD负相关;并且Zhang等近期证实,ucOCN通过GPRC6A受体保护小鼠免受高脂饮食诱导的NAFLD。然而,OCN对鸡肝组织的影响研究甚少,仅Wu等报道了注射ucOCN可减轻蛋鸡FLHS的胰岛素抵抗、肝组织炎症并可能激活肝组织自噬。另外,Jiang等研究显示,高能饮食可诱导蛋鸡脂肪肝、龙骨OCN浓度显著降低、骨转换上调导致骨骼损伤;因此,较低的OCN浓度也可能是骨质疏松和脂肪肝的联系因素之一。
1.5 有毒有害物质
饲料添加剂使用不当或饲料加工、生产、运输及存储环节产生有毒有害物质,如致病菌、黄曲霉菌、农药残留、重金属污染以及抗生素等易对肝造成损伤。致病菌如多杀性巴氏杆菌导致鸡肝细胞炎症等损害;肝为解毒器官,残留的农药使肝超负荷工作分解毒素,易引起肝纤维化等损害;同样,抗生素等药物不仅对肠道微生物群有影响,药物中成分也会对肝产生一定的毒性作用,进而造成损伤。肝损伤导致肝功能下降,从而可能使鸡易感FLHS。日粮中添加黄曲霉素B1导致肉鸡肝脂肪变性,引起PPAR信号通路失调导致蛋鸡肝脂肪沉积,并通过损伤线粒体结构和呼吸功能导致氧化应激、抑制抗氧化系统和细胞自噬、激活凋亡途径并通过miRNA影响细胞周期和生长、上调NF-κB炎症通路和炎症小体组装加剧肝毒性。环境中广泛存在的重金属镉等为不可生物降解的污染物,Go等报道称,低剂量镉暴露引起脂肪酸代谢失调导致小鼠出现脂肪肝,并刺激JNK磷酸化;氯化镉诱导增加了大鼠脂肪合成(SREBP1、SREBP2、FAS)基因和蛋白的表达,并提高血清和肝TG、TC水平导致肝脂质积累;同样,低剂量镉暴露上调蛋鸡肝脂质合成()和脂蛋白合成()基因表达,高剂量镉暴露产生ROS诱导氧化应激并引发肝损伤。
1.6 肠道微生物群:肠-肝轴
肠道屏障功能障碍、肠道微生物群及其代谢产物的变化影响NAFLD发生和进展。高脂饮食导致菌群失调,肠道屏障破坏,进而促使细菌或其产物通过门静脉转移至肝,肠道微生物对肝脂质代谢的影响如下:1)微生物失调导致肠道乙醇产量增加,乙醇可通过破坏紧密连接破坏肠道通透性,并诱导TG在肝内蓄积及肝氧化应激;2)肠源性病原体相关分子模式如LPS可特异性地与肝Toll样受体结合激活肝促炎症途径;3)肠道微生物增加胆碱的水解,进而导致胆碱缺乏,阻止VLDL分泌,导致TG在肝蓄积;4)过量的短链脂肪酸是肝糖异生和脂肪合成的底物,通过抑制AMPK活性促进肝FFA的积累。Li等报道,与未发生肝脂肪变性的蛋鸡相比,脂肪肝蛋鸡肠道中的条件致病菌和有害菌增多,有益菌减少,肝脂肪变性与细菌相对丰度有关。Hamid等用蛋鸡做模型研究自然发病NAFLD(FLHS)时发现,肠道中厚壁菌门的丰度与肝纤维化程度相反,而拟杆菌门丰度则与肝纤维化正相关,表明盲肠菌群失调与纤维化和NAFLD(FLHS)严重程度有关。在饲养过程中,给鸡添加益生菌可调节盲肠菌群,减少肝中的脂肪含量。Zhang等给肉鸡饲喂含有双歧杆菌、植物乳杆菌、粪肠球菌、丁酸梭菌、瓜尔胶的复合益生菌发现,该膳食补充剂改变了盲肠微生物群的多样性,改变了短链脂肪酸(short-chain fatty acids,SCFAs)的含量,抑制了脂肪生成;体外试验显示,SCFAs(包括醋酸盐、丙酸盐和丁酸盐)通过MAPK途径上调肠上皮细胞上胰高血糖素样肽-1(glucagon-like peptide,GLP-1)的表达,而GLP-1通过AMPK/ACC信号传导抑制原代肝细胞中的脂质积累。这些研究表明,与哺乳动物类似,鸡也具有肠-肝轴,FLHS疾病发展与肠道微生物密切相关。
2 FLHS模型的建立
鸡FLHS在笼养蛋鸡中尤为高发,严重影响养殖业健康发展,建立动物模型有利于对发病机制进行深入的研究,为防治该病奠定理论基础。且国内外研究表明,鸡FLHS与哺乳动物NAFLD相似,鸡FLHS常作为NAFLD的动物模型。饲料中能量含量过高是FLHS发生的主要原因之一,研究早期使用高能(high-energy,HE)日粮与强制喂食相结合可构建FLHS模型,为了更加符合养殖生产条件下的发病情况,后多为随意进食模型。现研究发现,高能低蛋白(high-energy low-protein,HELP)可能是鸡FLHS自然发生的原因之一,饲喂SFA来源的HELP非强制性进食模型成为建造鸡FLHS的经典方法。针对不同影响因素引起的鸡FLHS,临床上也使用甲硫氨酸/胆碱缺乏(MCD)日粮、肌肉注射雌二醇等方法诱导鸡FLHS模型(表1)。
2.1 高能(HE)日粮
能量过剩的蛋鸡有患FLHS风险,能量来源无论是碳水化合物还是脂肪均可诱发FLHS。1)碳水来源:饲喂高能玉米粮比低能大麦粮产生更高的FLHS发病率;2)脂肪来源:多项研究表明与碳水化合物相比,以脂肪为能量来源的平均出血评分较高,且以添加含SFA的动物油(猪油、牛油、羊脂)或提高n-6 PUFA比例(大豆油、菜籽油)更为明显。
2.2 高能低蛋白(HELP)日粮
Rozenboim等提出,HELP可能是诱导鸡FLHS的自然病理方式,商业养殖倾向于在鸡生长后期增加饲料能量并降低蛋白质含量,饮食中蛋白质含量不足以提供载脂蛋白合成所需的氨基酸,就会发生自然的FLHS。HELP日粮引起鸡高脂血症、肝空泡化、IL-1和TNF-α升高,导致肝胰岛素抵抗、脂肪变性和炎症,并且HELP日粮诱导比HE出血评分更高。
2.3 胆碱缺乏日粮
饲喂含有高能量、低亚油酸和低胆碱日粮的母鸡具有较高的肝重量和出血评分。MCD日粮导致50%鸡形成FLHS,但不同品系鸡均表现为HE比MCD日粮有着更高的发病率。最新研究显示,高胆固醇低胆碱(CLC)日粮和低蛋白高脂高胆固醇低胆碱(LPHFCLC)日粮促进鸡肝脂肪生成并导致脂肪性肝炎,7周龄雌鸡诱导4周可导致FLHS,该模式可用来快速生成研究脂肪变性和脂肪性肝炎的模型。
2.4 肌肉注射E2
Shini等使用30周龄蛋鸡肌肉注射E,以5 mg·kg剂量每4 d注射1次,诱导20 d可导致鸡的肝脏指数、肝脂肪含量、TG、TC显著增加,肝组织炎性细胞浸润并出血破裂,诱发FLHS。且该实验室进一步研究发现,E与LPS结合会增加FLHS的发病率和严重程度。然而,在另一项类似的研究中,E诱导了老年蛋鸡的FLHS,但不引起36周龄蛋鸡的FLHS。因此,该方法有待进一步确定功能性。
3 鸡FLHS的发生机制
FLHS的发生原因、机理和病理症状与哺乳动物NAFLD类似,但NAFLD发病机制极其复杂,目前尚未被完全揭示。早期学者普遍认可NAFLD和FLHS的发病机制是以胰岛素抵抗为第一次打击核心、氧化应激为第二次打击核心的“二次打击”学说,但随着深入研究,NAFLD的发病机制已从经典“二次打击”学说演化为“多重打击”。“多重打击”对研究鸡FLHS的发病机制也具有重要参考意义。鸡由于其自身生理特性和外部高脂饮食易形成高血糖、高血脂症,产生胰岛素抵抗。胰岛素抵抗和高脂饮食共同使脂质代谢异常,脂肪以甘油三酯的形式在肝中积累诱发单纯性脂肪变性;与此同时,脂质代谢异常产生的高水平FFA、游离胆固醇和脂质中间体导致脂肪毒性增强,并进一步促进胰岛素抵抗。因此,胰岛素抵抗是引发鸡FLHS的第一因素。脂肪毒性使内质网应激和线粒体功能障碍机制被激活,由此产生过量ROS导致氧化应激,而胰岛素抵抗、脂肪毒性、氧化应激和胰岛素抵抗引起的脂肪因子分泌异常等同时或级联地进一步激活肝组织炎症、损伤和纤维化、自噬抑制,而以上的“多重打击”最终导致鸡FLHS。
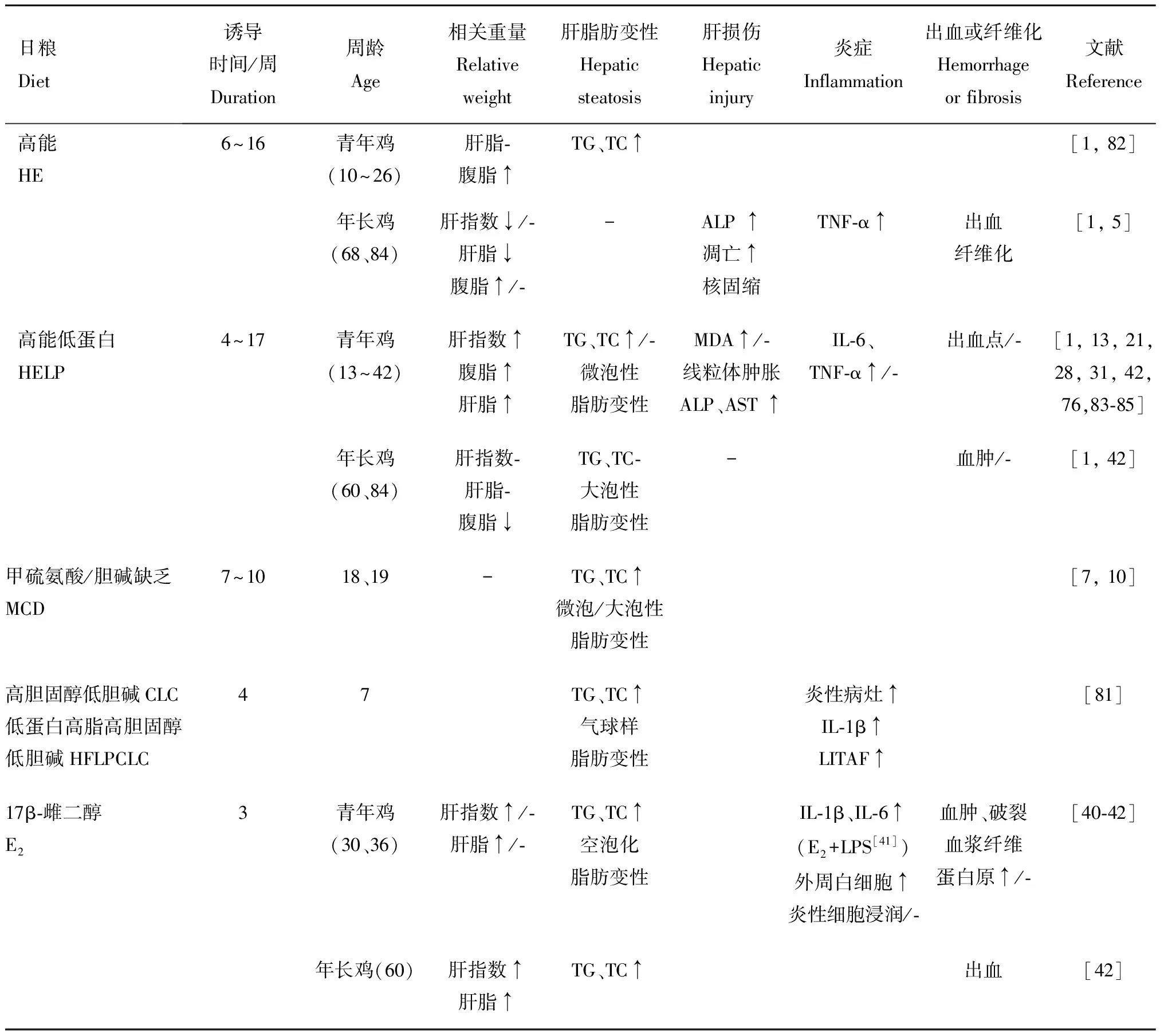
表1 近5年鸡FLHS诱导模型及相关指标变化
3.1 肝游离脂肪酸增多
鸡肝脂肪合成速度远胜于哺乳动物,且肝外组织合成脂肪能力有限,超过90%的脂肪在肝内合成。鸡的淋巴系统不够完善,从肠道吸收的脂肪酸通过肝门静脉系统直接转运至肝,肝合成的脂肪和蛋白结合形成肝脂蛋白,大部分以VLDL的形式转运到其他组织中储存或利用,如发育的卵母细胞以形成蛋黄。当肝DNL和通过肠道吸收的脂肪酸高于脂肪酸β氧化和VLDL分泌能力时,过量FFA和TG在肝中积累,造成单纯性脂肪变性。
3.1.1 肝脂肪从头合成增强 转录因子如SREBP-1、ChREBP和PPARγ的激活可增强肝DNL过程。ACC和FAS是DNL过程中脂肪酸生成的关键酶,临床上肝靶向ACC抑制剂减少非酒精性脂肪肝炎(nonalcoholic steatohepatitis,NASH)患者肝脂肪含量,FAS抑制剂降低代谢异常男性的DNL。LXRα为调节肝脂质代谢的转录因子,在NAFLD中被激活促进SREBP-1c转录,提高ACC和FAS活性,阻断LXRα磷酸化延缓高脂高胆固醇饮食小鼠的NAFLD进展。因此,肝DNL异常增强是NAFLD发展的重要途径。同样在鸡中,不同日粮诱导的FLHS均表现为肝中SREBP-1c、PPARγ、ACC和FAS显著上调,DNL增强,TG和FFA含量增多。
3.1.2 肝脂肪酸β氧化减少 肉碱棕榈酰转移酶(carnitine palmitoyl transterase,CPT)是线粒体脂肪酸β氧化的关键酶,丙二酰CoA作为DNL合成早期的中间体,可抑制CPT活性。PPARα和能量传感器AMPK信号通路为脂肪酸氧化的积极调节因子,PPARα和AMPK信号通路失调导致脂肪酸β氧化能力的降低,影响FLHS的发生。PPARα可上调CPT-1表达,增强线粒体和过氧化物酶体的脂肪酸氧化能力,也可通过增强细胞色素P450表达促进微粒体ω氧化,并且PPARα还可抑制炎症和急性期反应;鸡FLHS表现为肝组织中PPAR信号通路失调,使用PPARα激动剂则降低肉鸡基因和蛋白水平表达,进一步抑制SREBPs转录、降低TG和TC含量。Gao等报道FLHS蛋鸡1基因表达水平显著下调;Zhang等使用AMPK激动剂则显著上调蛋鸡肝细胞、-1基因水平,下调、基因水平,表明AMPK可减少脂质合成,促进脂肪酸氧化。3-羟基丁酸为脂肪酸β氧化的特定代谢物,Guo等研究发现,蛋鸡FLHS血清3-羟基丁酸含量显著降低,Feng等研究发现,肝PPARα的激活能促进脂肪酸利用,并增加血清3-羟基丁酸的含量。此外,鸡FLHS的肝中柠檬酸合酶和细胞色素C氧化酶的活性降低,另一项研究中,表现为-1、和(甘油三酯水解标志酶)基因下调。肝脂肪酸β氧化减少引起了脂肪酸的过度积累。
3.1.3 脂滴的形成 过多脂肪酸在肝中形成TG并以脂滴(lipid droplets, LDs)形式暂时储存或以含TG的极低密度脂蛋白(VLDL-TG)形式分泌。鸡FLHS中大部分表现为肝TG增多和脂滴积累导致肝细胞不同程度空泡化(脂肪变性)。Apo B定位在内质网膜启动VLDL囊泡组装, MTTP作为关键酶参与了Apo B在内质网的转运以及转移脂肪到新生脂蛋白颗粒中,两者是VLDL组装和分泌过程中的重要蛋白;功能失调的VLDL-TG合成和分泌是单纯性脂肪变性转变为脂肪性肝炎的原因,阻断Apo B、MTTP和VLDL分泌导致NASH/NAFLD。肝MTTP在FLHS鸡中表达增加,表明TG在VLDL中积累;Song等研究发现,鸡肝中Apo AⅠ和Apo B100在FLHS鸡中异常降低,表明VLDL的功能失调是FLHS的一部分,VLDL分泌受阻又会进一步导致LDs的形成,导致肝细胞脂肪化。
3.2 胰岛素抵抗
鸡肝组织中,胰岛素与胰岛素受体结合,使其自磷酸化并磷酸化胰岛素受体底物(insulin receptor substrate,IRS)的酪氨酸残基以启动胞内胰岛素信号转导,即PI3K/Akt和MAPK ERK1/2两条主要转导途径。已证实鸡肝中存在的IRS包括IRS-1和Shc(主要为52 ku亚型),虽然鸡基因组1号染色体存在IRS-2同源物编码序列,但IRS-2尚未在鸡各组织中报道。Dupont等研究发现,胰岛素级联反应在鸡肝中正常表现,但在鸡脂肪和肌肉组织存在明显的胰岛素抵抗(insulin resistance, IR),具体表现为胰岛素刺激或抑制不影响胰岛素受体、IRS-1、Shc的酪氨酸磷酸化和下游元件(PI3K、Akt、AMPK)的活性,具有天然的“胰岛素抵抗症”。胰岛素通过葡萄糖转运蛋白4(GLUT4)增强对葡萄糖的摄取,而在鸡组织中缺乏GLUT4,因此鸡的血糖浓度水平是其体重相近哺乳动物的2~4倍,其生理特征较哺乳动物更易诱发IR。研究显示,FLHS鸡空腹血糖和TG浓度显著增加,产生高水平IR,表明IR参与了鸡FLHS。
3.2.1 FFA诱导促进胰岛素抵抗 FFA引起IR的机制包括细胞内脂质中间体、内质网应激/氧化应激、促炎细胞因子的产生。一方面,大量FFA及其脂质中间体如二酰基甘油(DAG)、脂酰CoA和神经酰胺等具有脂肪毒性,导致线粒体功能障碍(氧化活性和ATP合成降低),抑制胰岛素分泌(胰岛素分泌消耗ATP);DAG和内质网应激/线粒体功能障碍产生的过量ROS可激活蛋白激酶C(PKC),而PKC可降低IRS-1/2的酪氨酸磷酸化。另一方面,FFA介导NF-κB的激活和促炎细胞因子产生(IL-1β、IL-6、TNF-α),促炎细胞因子激活JNK1和细胞信号转导抑制因子(SOCS),二者增强IRS1/2丝氨酸磷酸化,抑制酪氨酸磷酸化。因此,FFA通过多种途径导致胰岛素信号通路缺陷,产生IR。
3.2.2 胰岛素抵抗加剧FFA的生成 IRS激活可作为SREBP-1c的调节因子,IR状态下,IRS下调使SREBP-1c过度表达进而驱动肝DNL。正常情况下,胰岛素与其受体结合使参与TG水解为甘油和FFA的激素敏感酶(hormone-sensitive lipase,HSL)失活,抑制TG分解;而在IR状态下,HSL将FFA释放到肝循环中的活性增强,从而进一步引起肝FFA水平大量增加,造成恶性循环。
过量FFA与IR的恶性循环是导致鸡FLHS的重要诱因之一,并且两者共同导致脂肪因子分泌异常,并激活脂肪毒性、氧化应激和炎症的级联反应。
3.3 脂肪毒性、氧化应激
脂肪毒性与氧化应激的恶性循环加重鸡FLHS,且进一步激活炎症信号通路。
3.3.1 内质网应激 正常含量的脂质成分控制内质网、线粒体的结构和功能,这取决于内质网运输到线粒体的脂质数量、线粒体合成磷脂的能力。磷脂酰丝氨酸(phosphatidyl serine,PS)主要在内质网合成,通过线粒体相关内质网膜(mitochondria-associated endoplasmic reticulum membranes,MAMs)导入线粒体转化为磷脂酰乙醇胺(phosphatidyl-ethanolamine,PE),PE再导入内质网转化为磷脂酰胆碱(phosphatidylcholine,PC)。研究表明,MAMs的完整性决定了内质网-线粒体的正常通讯,其被脂质破坏时可损害内质网Ca稳态、导致内质网应激(endoplasmic reticulum stress,ERS),靶向MAMs完整性的药物可改善NAFLD。线粒体融合蛋白2(mitofusin-2,Mfn2)为线粒体膜蛋白,具有连接内质网膜和线粒体的作用,Mfn2结合PS并特异性地将PS提取到膜区转移至线粒体。Hernández-Alvarez等最新研究发现,在NASH患者和小鼠模型中均检测到Mfn2水平降低,减少了内质网PS转移和PC合成,导致ERS,并提出破坏内质网-线粒体PS转移可能是肝疾病发展的新机制。细胞色素P4502E1(CYP2E1)为内质网膜末端氧化酶,Ca稳态破坏、ERS提高CYP2E1活性而产生过量ROS,导致氧化应激。另一方面,蛋白质合成增加、ERS或缺乏ATP导致内质网内未折叠蛋白质的积累,激活的“未折叠蛋白反应 (unfolded protein response,UPR)”是解决ERS的适应性反应。在NAFLD中,诱导UPR的因素包括高血糖、高胆固醇血症、线粒体损伤、ATP缺乏、PC耗竭和氧化应激。在长期或严重的ERS下,UPR由适应性转变为激活JNK1信号通路和线粒体凋亡途径启动促凋亡机制。Gesek等在研究Ross 308、Cobb 500和 Hubbard F153 三个品种肉鸡育肥过程中肝组织超微结构变化时发现,在育肥的17、31、38 d随着肝细胞细胞质显示大小不等的空泡或脂滴,线粒体发生肿胀、变形、增殖和损伤,粗面内质网出现碎片化和腺泡转化。内质网应激可能参与了禽FLHS发展,但需要进一步开展更多的研究来证实。
3.3.2 氧化应激 大量脂质中间体如脂酰CoA进入线粒体内膜进行β氧化,增加的脂质流量使呼吸链崩溃,增加电子泄露使ROS过量产生,导致氧化应激。大量FFA、脂质中间体、ROS以及ROS氧化生物膜产生的脂质过氧化物等具有脂肪毒性,改变细胞膜流动性和通透性,造成线粒体Ca内流增加、结构改变和功能障碍,进一步产生ROS形成氧化应激增强的“恶性循环”,并且氧化应激通过上调肝DNL进一步驱动脂肪变性。在鸡FLHS进程中,Xing等报道高能低蛋白导致蛋鸡肝中丙二醛(malondialdehyde,MDA)浓度显著增加,而超氧化物歧化酶、过氧化氢酶和谷胱甘肽水平显著降低;同样,Wu等报道高脂日粮诱导FLHS鸡的肝中MDA浓度升高,GSH-Px降低,造成氧化应激。
3.4 炎症和纤维化
3.4.1 NLRP3炎症小体和IL-1β NLRP3炎症小体是炎症反应中重要的组成部分,为大型细胞质多聚体复合物。脂质积累使细胞受损/死亡释放ATP,胞外高浓度ATP可激活靶细胞中嘌呤能受体(如P2X7受体),受体激活NLRP3炎症小体组装,并诱导IL-1β成熟和分泌,加强炎症反应。NLRP3的磷酸化促进NLRP3炎症小体复合物的组装,而JNK1可介导NLRP3磷酸化;线粒体功能障碍、ROS产生以及溶酶体损伤进一步促进磷酸化NLRP3炎症小体自身结合和随后的炎症体组装。NLRP3炎症小体缺陷小鼠全身神经酰胺水平降低,炎症减轻,且免受饮食诱导NASH疾病的进一步发展。炎症小体激活还促进肝纤维化,3基因敲入小鼠在正常饮食下表现出自发性肝纤维化,Gaul等最新研究发现,肝细胞外NLRP3炎症小体可被肝星状细胞吞噬,使IL-1β 和α-平滑肌肌动蛋白表达增加,导致肝纤维化,揭示细胞间炎症传播的新方式。与单纯脂肪变性患者相比,NASH患者NLRP3和前IL-1β的表达显著升高,并且IL-1β进一步加强TNF-α诱导的细胞损伤和炎症,推动NASH发展。降低鸡只NF-κB/NLRP3/Caspase1相关通路表达能减轻肝炎症反应和细胞焦亡,但是,NLRP3炎症小体与鸡FLHS关系研究甚少。
3.4.2 炎症状态 过量的FFA直接刺激TNF-α的表达,TNF-α和ROS主要通过刺激NF-κB和JNK1/2炎性通路使促炎细胞因子(IL-1β、IL-6、TNF-α)进一步产生和释放。与单纯脂肪变性患者相比,NASH患者血清TNF-α水平更高,且肝组织TNF-α和TNF受体1(TNFR1)的表达水平与脂肪性肝炎进程和纤维化严重程度相关。TNF-α介导的肝损伤主要通过TNFR1转导,Wandrer等研究表明,在NAFLD小鼠模型中选择性抑制TNFR1可抑制SREBP1及其下游和JNK的激活,抑制肝脂肪变性、炎症性损伤、胰岛素抵抗和纤维化。然而,TNFR1相关功能在鸡FLHS中尚无报道。NF-κB是炎症激活的主要转录因子,在NAFLD动物模型以及NASH患者中显示NF-κB的持续激活,而其持续激活导致慢性炎症状态。JNK1/2是炎症和凋亡的激活剂,抑制JNK减少脂肪变性、脂肪性肝炎和胰岛素抵抗。如前所述,TNF-α、NF-κB和JNK1/2都可导致胰岛素抵抗和炎症状态,因此,炎症信号通路和促炎因子持续激活在脂肪变性发展为脂肪性肝炎和肝纤维化过程中起关键作用,且导致脂肪性肝炎/纤维化的常见组织学变化,如细胞凋亡和坏死、中性粒细胞浸润、肝星状细胞活化和mallory小体产生。最近有研究检测到FLHS鸡的肝中IL-1β、TNF-α表达升高,导致脂肪性肝炎;FLHS鸡的肝脂肪变性和局部出血并伴随淋巴细胞、单核细胞和嗜异性粒细胞浸润,肝-1、-6基因表达增高、外周白细胞总数和纤维蛋白原水平升高,表明肝局部和全身性炎症引起损伤并最终导致鸡FLHS。
因此,FFA水平的增加以及随之而来的胰岛素抵抗、脂肪毒性、氧化应激、炎症等促发一系列级联反应,导致肝的炎症、损伤和纤维化。
3.5 脂肪因子分泌异常
脂肪组织被认为是控制新陈代谢的激素活性系统,可分泌激素和脂肪因子,如瘦素(leptin)和脂联素(adiponectin,ADPN),影响葡萄糖稳态、脂质代谢和炎症。脂肪组织中过多的脂质储存导致胰岛素抵抗、NAFLD和其他脂质代谢疾病,而胰岛素抵抗进一步导致脂肪因子失衡,不仅严重影响脂肪组织本身,还会影响肝的脂质代谢。
瘦素是脂肪细胞分泌的重要脂肪因子,可调节脂肪储存,加快生物的新陈代谢,抑制食欲,具有促炎作用,并防止非脂肪部位的脂质积聚。随着研究的不断深入,发现其受体不仅存在于丘脑、脂肪组织,还广泛存在于全身各个组织如乳腺上皮细胞、胎盘、胃黏膜上皮细胞。在哺乳动物肝中,瘦素通过降低SREBP-1的表达调节脂质代谢;通过hedgehog和m-TOR途径激活肝星状细胞,其促纤维化作用已在各种体外和动物模型中得到证实;瘦素还具有激活肝细胞自噬的功能,血清瘦素水平对NAFLD的作用和意义仍未明确。鸡瘦素基因由于低序列保守性、低表达水平,GC含量高达70%,直到2016年才被确认,且目前暂无瘦素对鸡肝的影响和机制研究。Seroussi等研究明确了鸡瘦素在肠道黏膜上皮柱状细胞和杯状细胞中表达。因此,不同于哺乳动物中瘦素作为循环激素,鸡瘦素可能通过自分泌/旁分泌模式作用,通过肠-肝轴影响鸡FLHS。
ADPN作为一种胰岛素超敏化激素,可改善肝和外周胰岛素抵抗,并具有抗炎和保肝活性。ADPN水平与代谢综合征和肝功能障碍的风险呈强负相关,其在肥胖中的水平降低可能与线粒体功能障碍和胰岛素抵抗有关。ADPN还具有直接的抗纤维化作用,这可能通过激活脂联素受体1(AdipoR1)-AMPK途径介导;在缺乏ADPN的小鼠中已证实其肝纤维化增强,而注射重组脂联素可显著改善小鼠的脂肪性肝炎和纤维化。最近,新型合成活性小分子脂联素受体激动剂(AdipoRon)在体内外显示出与ADPN相似的作用,AdipoRon通过AdipoR1-AMPK和AdipoR2-PPARα途径发挥抗氧化和抗炎作用。ADPN和AdipoR1/2可在鸡肝中较高表达,不同于哺乳动物的ADPN为3种不同的寡聚体,鸡ADPN结构为独特的脂联素聚合物,其质量大于669 ku。然而,ADPN对鸡肝的作用暂无报道,有待研究。
3.6 自噬抑制
自噬调节脂质代谢和胰岛素抵抗,清除异常线粒体保护肝细胞免受损伤和死亡,且多方证据支持自噬抑制在NAFLD进程中起核心作用。脂噬为自噬的一种,其介导的LDs脂肪分解为较新发现,Gao等报道高脂饮食抑制小鼠脂噬,而运动和饮食干预分别通过激活AMPK/ULK1和抑制Akt/mTOR/ULK1途径增强肝的脂噬进而改善NAFLD;沉默自噬相关基因(5、7)表达或抑制剂阻断自噬导致LDs积累,而通过肝特异性过表达Atg7、FOXO1、TFEB等或使用自噬激活剂则可降低肝脂肪变性和胰岛素抵抗。自噬清除ROS和受损线粒体DNA从而抑制炎症小体激活,小鼠肝细胞特异性5基因敲除通过IL-1β/TNF途径导致肝组织炎症增加;NAFLD患者肝62/1基因和蛋白表达增高,其积累代表自噬抑制,并与纤维化阶段显著正相关。然而,自噬在肝癌发生发展的不同阶段其影响呈双面性,自噬抑制肝癌的发生和调节免疫发挥抗肿瘤作用,一旦肿瘤形成,自噬则通过降解受损细胞器和异常蛋白提供营养使癌细胞得以存活进而转为促进肝癌发展。诱导RAW264.7细胞脂肪酸氧化抑制细胞自噬;Wang等通过高能低蛋白日粮诱导的FLHS蛋鸡,发现肝自噬相关基因-1、5和7表达量降低,62基因表达量升高。Wu等利用高脂日粮诱导FLHS蛋鸡,发现FLHS蛋鸡肝自噬小体和自噬溶酶体数量减少。以上研究均表明自噬抑制参与了鸡FLHS的发展,但自噬如何调节FLHS还有待进一步的研究。
4 总结和展望
受遗传、表观遗传修饰、营养、激素和环境等多重影响因素使鸡易感FLHS,而游离脂肪酸增多、胰岛素抵抗、脂肪毒性、氧化应激、炎症、纤维化、脂肪因子分泌障碍和自噬抑制等“多重打击”导致积累性肝损伤最终引起鸡死亡。对于模型的选择,通过对近5年鸡FLHS模型的分析总结出:1)诱导日粮的选择:高能低蛋白(HELP)日粮>高能(HE)日粮>甲硫氨酸/胆碱缺乏(MCD)日粮,如需快速获得鸡FLHS可将几种模型结合。2)鸡年龄的选择:年长鸡比青年鸡更加易感FLHS,青年鸡易诱导FLHS进程前期的脂肪变性和炎症模式,年长鸡易诱导FLHS进程后期的出血和纤维化模式。
鸡FLHS的形成具有多重影响因素,表现为系统间的平衡至关重要;鸡日粮中的各种成分通过肠-肝轴发挥作用、生殖系统中雌二醇的变化、应激引起肾上腺相关激素的改变、骨组织分泌的骨钙素以及脂肪因子脂联素等能量调控因子的变化、鸡脂肪和肌肉组织本身存在明显的胰岛素抵抗,体现出机体维持健康的基础是各器官系统良性的动态平衡。目前,鸡FLHS“多重打击”机理研究较少,在脂肪酸、甘油三酯、胰岛素抵抗和炎症因子方面较多,但瘦素、脂联素、炎症小体和自噬在FLHS发展中的作用还未明确;瘦素由于其特殊的表达位置可能通过肠-肝轴影响FLHS,已报道FLHS进展中存在自噬但具体作用有待确定。由于鸡FLHS临床症状表现不明显,发病突然死亡,临床应多以预防为主,可针对不同的影响因素和作用机制筛选药物,利用营养补充剂、骨钙素、益生菌、益生元、合生元、粪菌移植以及信号通路相关的siRNA、miRNA、脂联素受体激动剂、AMPK激动剂、自噬激动剂、JNK抑制剂等有望预防和治疗鸡FLHS。
猜你喜欢
中国现代医生(2022年20期)2022-11-04
中国现代医生(2022年21期)2022-08-22
中国药学药品知识仓库(2022年9期)2022-05-23
家庭医学·下半月(2022年3期)2022-04-07
家庭医药·快乐养生(2020年10期)2020-11-06
健康之家(2020年7期)2020-11-02
家庭科学·新健康(2020年6期)2020-07-06
科学导报(2019年45期)2019-09-23
保健与生活(2019年23期)2019-09-10
中国医药导报(2019年13期)2019-06-20
