
固相萃取—超高效液相色谱—质谱法测定食用香精中3种甲基咪唑类物质
2022-11-02 01:08周艳华徐文泱
食品与机械 2022年10期
周艳华 李 涛 向 俊 徐文泱
食品中常见的美拉德反应可产生赋予食品色、香、味的成分,同时也可能伴随产生甲基咪唑类化合物[1-2]。甲基咪唑是一类五元杂环化合物,对动物有强烈惊阙作用,可诱发癫痫。目前,甲基咪唑化合物检测主要有紫外分光光度法[3]、气相色谱法[4]、液相色谱法[5]、质谱法[6-9]等。紫外分光光度法成本低,操作简单,但灵敏度低,适用范围小,且易出现假阳性结果。气相色谱法定量准确,适用于易挥发、成分简单样品,但对于复杂样品,易出现假阳性,重复性差。液相色谱法定量准确,但灵敏度不高,前处理需衍生化,操作步骤复杂。气相质谱法易受杂质干扰,出现假阳性结果,不能满足痕量测定需求。液质法选择性强,灵敏度高,被广泛应用于食品中痕量有毒有害物的检测,是检测甲基咪唑的最优方法。甲基咪唑分子量较小,质谱裂解可能会出现假阳性和基质效应,内标法是保障结果准确性的有效手段。
现有研究主要集中于含焦糖色素和添加焦糖色素的食品(如酱油[10]、耗油[11]、豆制品[12])中甲基咪唑的检测,多采用GB 5009.282—2020方法,此法不适用于食用香精等食品添加剂检测。研究拟使用三氯乙酸高效提取除杂,采用固相萃取法(SPE)和超高效液相色谱与质谱联用法(UPLC-MS/MS)进行富集净化和定性检测,最后利用内标法进行定量分析,旨在建立一种快速检测食用香精中甲基咪唑、2-甲基咪唑和4-甲基咪唑的方法,为食品安全监管部门有效监控食用香精中3种甲基咪唑化合物的来源提供技术支持。
1 材料与方法
1.1 材料与试剂
麻辣香精、牛粉香精、鸡肉膏等:市售;
乙腈、甲醇:色谱纯,德国默克公司;
三氯乙酸、乙酸铵、氨水:优级纯,上海国药试剂有限公司;
1-甲基咪唑:纯度>99%,上海安谱实验科技股份有限公司;
2-甲基咪唑:纯度>99%,北京坛墨质检科技有限公司;
4-甲基咪唑:纯度>98.5%,北京曼哈格生物科技有限公司;
1-甲基咪唑-D6、2-甲基咪唑-D6内标物:纯度≥98%,天津阿尔塔科技有限公司;
4-甲基咪唑-D6内标物:纯度>98%,加拿大CDN公司。
1.2 仪器与设备
质谱仪:TSQ Quantis型,美国赛默飞公司;
分析天平:BSA224s型,赛多利斯科学仪器(北京)公司;
冷冻离心机:AVANTI J-15R型,贝克曼库尔特公司;
超声波清洗器:SCQ型,上海声彦超声波仪器有限公司;
数显型多管旋涡混合器:945066型,美国Talboys公司;
高速振荡器:CM-1000型,东京理化器械株式会社。
1.3 方法
1.3.1 标准溶液配制 分别精密称取1-甲基咪唑、2-甲基咪唑、4-甲基咪唑、1-甲基咪唑-D6、2-甲基咪唑-D6、4-甲基咪唑-D6标准品,用乙腈溶解并配制成质量浓度为1.0 mg/mL的标准储备液,使用前稀释为1.0 μg/mL的标准使用液。分别精密移取一定量的标准使用液,加入同位素内标溶液,配制成标准线性系列溶液,质量浓度为5.0~400.0 ng/mL,内标质量浓度为50.0 ng/mL。
1.3.2 样品前处理 精密称取混匀后的食用香精2 g于离心管中,加入1.0 μg/mL的混合内标溶液0.25 mL和25 mL 5%三氯乙酸溶液,振摇20 min后,冷冻离心,精密量取5 mL上清液,过混合阳离子交换固相萃取小柱净化,依次用5 mL水溶液、5 mL正己烷和5 mL甲醇溶液淋洗,用8 mL 5%氨水—甲醇洗脱,氮气吹至近干,精密移取1 mL 5 mmol/L乙酸铵—乙腈(体积比10∶90)溶解,过膜,待测。
1.3.3 液相色谱条件及质谱条件优化 液相色谱条件为:A为乙酸铵溶液(5 mmol/L),B为乙腈,流速0.4 mL/min,柱温30 ℃,进样体积5 μL。梯度洗脱程序:0~4.5 min,10% A;4.5~4.6 min,10% A~40% A;4.6~5.9 min,40% A;5.9~6.0 min,40% A~10% A;6.0~8.0 min,10% A。在HESI离子源下,毛细管电压3 500 V,雾化器温度350 ℃,离子传输管温度350 ℃,鞘气4.58 L/min,辅助气7.97 L/min,反吹气1.5 L/min。通过标准物质母离子扫描和子离子扫描,确定质谱分析方法。
1.3.4 方法学验证 在空白香精样品中添加低浓度混合标准物质,再在确定的最优前处理条件和仪器条件下测定,S/N≥3的浓度为检出限,S/N≥10的浓度为定量限。在空白基质样品中加入3种甲基咪唑及内标,甲基咪唑含量为5.0,10.0,50.0 μg/kg(n=6),按确定的前处理工艺和仪器方法测定,计算回收率和相对标准偏差值。
2 结果与讨论
2.1 质谱条件优化
通过CAS号查询3种甲基咪唑及内标物的分子量,通过母离子全扫描结果可知3种甲基咪唑及内标响应度最高的是正离子模式。通过子离子扫描,获得目标化合物的最优质谱参数,甲基咪唑化合物依据响应强度确定定量离子和定性离子,3种甲基咪唑化合物的质谱检测方法见表1。3种甲基咪唑分子量较小,经质谱电离后产生的碎裂离子数较少,碎片离子质量小。选择稳定性较好、响应度较强和碎片离子质量较高的离子作为定量离子,可以提高检测的稳定性。
2.2 色谱条件优化
3种甲基咪唑化合物是同分异构体,分子量相同,裂解定量离子也相同,因此需优化色谱条件来分离3种甲基咪唑化合物。首先采用C18系列色谱柱,研究发现增加色谱柱长度对3种甲基咪唑出峰时间影响较小,出峰时间均在1 min左右,且3种甲基咪唑保留时间一致,再选用C8色谱柱,分离效果无改善。目标化合物出峰时间较早,易与样品基质同时出峰,影响分析结果的准确性。甲基咪唑化合物是含氮极性小分子化合物,C18或C8色谱柱对此类化合物保留能力弱,因此出峰时间早,不能有效分离3种化合物。因此选择保留能力与C18相反的亲水性色谱柱(HILIC)来分离3种甲基咪唑。研究选择了Dikma Inspire HILIC(2.1 mm×100 mm,3 μm)、Ultimate UHPLC HILIC(2.1 mm×150 mm,1.8 μm)和Waters ACQUITY UPLC BEH Amide HILIC(3.0 mm×150 mm,1.7 μm)在其最优的色谱条件下分离3种甲基咪唑,结果表明Waters ACQUITY UPLC BEH Amide HILIC 色谱柱能有效分离3种甲基咪唑化合物,其他两种色谱柱可将3种甲基咪唑化合物的保留时间延后,但不能将3种同分异构体化合物有效分离,因此选择Waters ACQUITY UPLC BEH Amide HILIC进行检测分析。此色谱柱填料为1.7 μm的BEH HILIC颗粒,具有高分离度、高柱效、高分析速度等特点,因此可以将3种同分异构体化合物进行分离,通过单一标准物质进样分析,出峰的顺序分别为1-甲基咪唑、4-甲基咪唑、2-甲基咪唑,3种甲基咪唑及其内标化合物定量离子色谱图见图1。
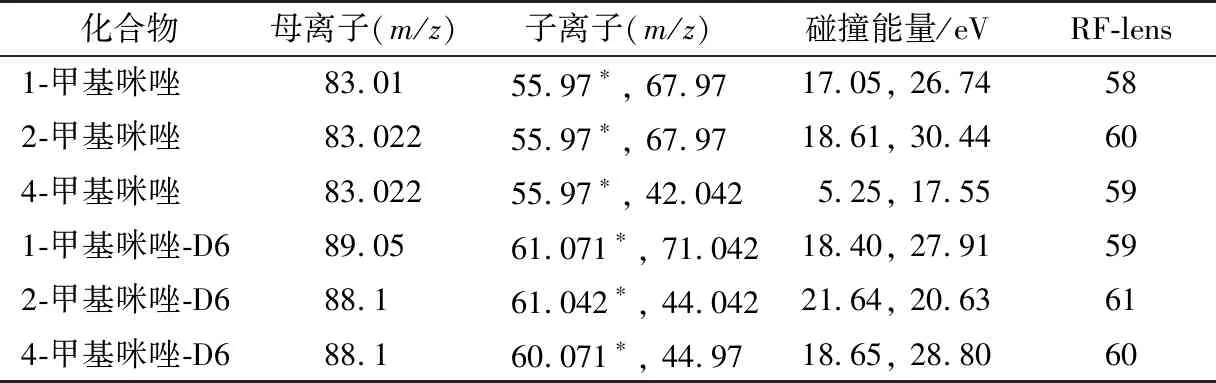
表1 甲基咪唑化合物的质谱参数表†Table 1 Mass spectrum parameters of methylimidazole compounds
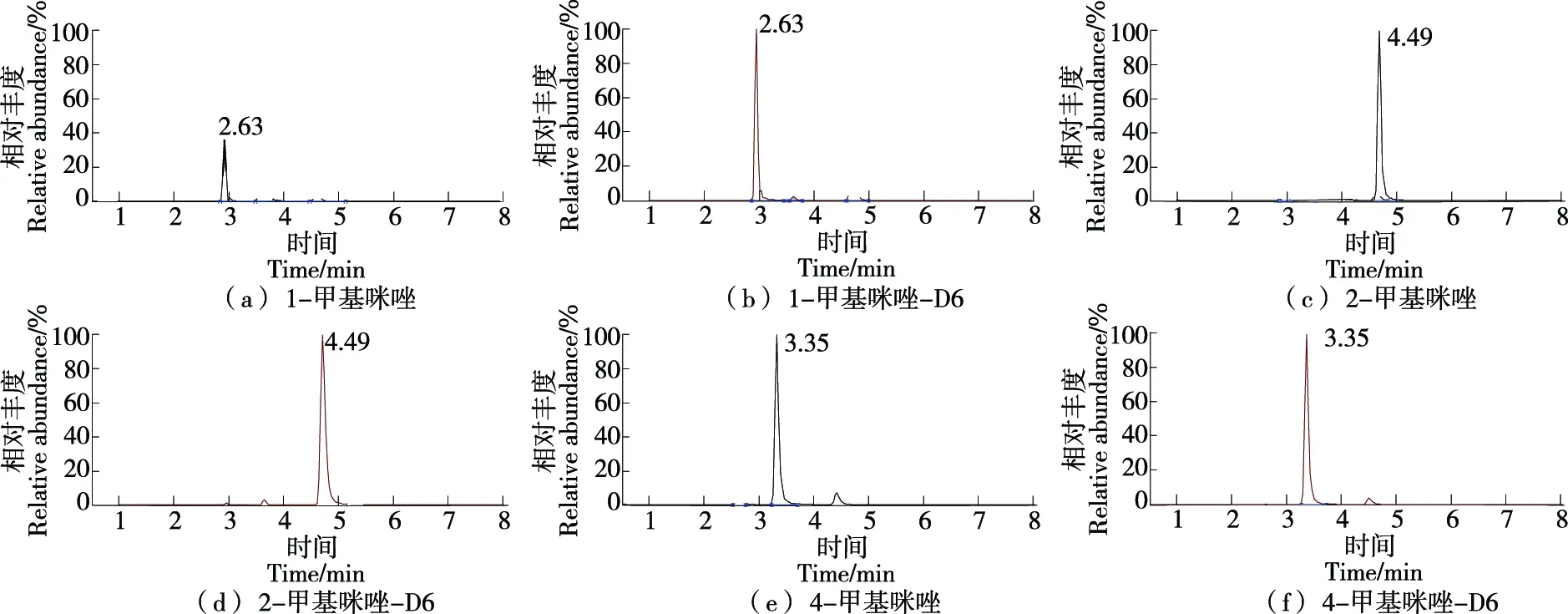
图1 3种甲基咪唑化合物及其内标混合标准溶液定量离子色谱图Figure 1 Quantitative ion chromatogram of three methylimidazole compounds and their internal standard mixed standard solutions
2.3 提取试剂的优化
甲基咪唑化合物易溶于水,因此选用水系溶剂进行提取。研究选择水、2%甲酸水溶液、2%三氯乙酸水溶液进行提取效果对比,结果见图2。由图2可知,3种提取试剂均能提取3种甲基咪唑化合物,当用水提取时,甲基咪唑化合物回收率最低,2%三氯乙酸的提取回收率最高。在净化过程中,水提取溶液过柱净化速度最慢,2%三氯乙酸提取溶液最快。结合提取回收率和净化效率,选择三氯乙酸溶液作为提取试剂。复合食用香精中含有肽、氨基酸、蛋白质、脂肪等,在提取甲基咪唑的前处理过程中,三氯乙酸可以有效沉淀蛋白质,提高提取效率。研究进一步比较了1%三氯乙酸溶液、2%三氯乙酸溶液、5%三氯乙酸溶液、10%三氯乙酸溶液的提取效果,见图2。由图2可知,随三氯乙酸提取液体积分数的升高,甲基咪唑化合物的回收率逐步升高,但当三氯乙酸体积分数≥5%时,甲基咪唑化合物的回收率变化较小。这是因为随三氯乙酸提取液体积分数的升高,提取剂中三氯乙酸沉淀杂质的能力越强,当三氯乙酸体积分数为5%时,样品已被处理澄清,达到最佳的净化效果,过量的三氯乙酸溶液会增加后续淋洗次数。因此,选择5%的三氯乙酸提取作为香精中甲基咪唑的提取液。
2.4 净化固相萃取柱的优化
在最优提取工艺条件下,比较了强阳离子固相萃取柱(SCX)、混合阳离子固相萃取柱(PCX)、弱阳离子固相萃取柱(WCX)对3种甲基咪唑的净化效果。不同固相萃取柱净化回收率见图3。结果表明,经PCX柱净化后,3种甲基咪唑化合物回收率最高,WCX固相萃取柱净化回收率最低。PCX柱利用阳离子交换机理提供了离子交换与反相保留双重保留模式,对碱性化合物具有高的选择性,而WCX柱填充了混合型弱阳离子交换反相吸附剂,对强碱性的化合物具有高选择性,适用的pH值范围比较窄,SCX柱填料键合基团通常为(苯)磺酸基,一般用于有机碱类化合物净化。3种甲基咪唑为含氮五元芳香杂环化合物,呈弱碱性,PCX柱对3种甲基咪唑进行选择性吸附,选择不同极性淋洗液淋洗净化,氨化甲醇洗脱回收目标物,达到富集目标化合物并消除杂质,降低杂质引起的基质效应。

图2 不同提取剂对甲基咪唑回收率的影响Figure 2 Effects of different extractants on the recovery of methylimidazole
2.5 线性关系、检出限和定量限
由表2可知,当质量浓度为5.0~400.0 ng/mL时,3种甲基咪唑化合物的线性关系良好,相关系数R2≥0.997,检出限(LOD)为0.030~0.45 μg/kg,定量限(LOQ)为0.10~1.5 μg/kg,此方法对3种甲基咪唑的检出限和定量限灵敏度高于GB 5009. 282—2020。3种甲基咪唑分子量相同,但分子结构不同,在同一质谱条件下响应度有差异,因此检出限和定量限也不同。
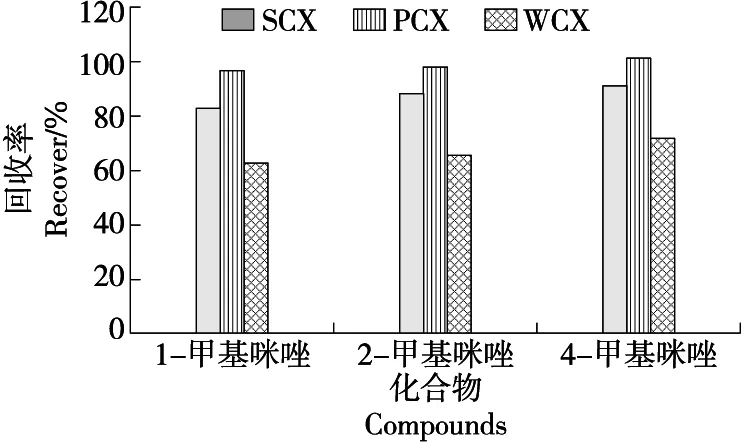
图3 不同固相萃取柱对甲基咪唑回收率的影响Figure 3 Effects of different solid phase extraction columns on the recovery of methylimidazole
2.6 准确度和精密度
由表3可知,当甲基咪唑化合物加标量为5.0 μg/kg时,回收率在82.8%~111.7%,相对标准偏差RSD为1.2%~6.6%;当甲基咪唑化合物加标量为10.0 μg/kg时,回收率在85.5%~105.2%,RSD为1.3%~9.3%;当甲基咪唑化合物加标量为50.0 μg/kg时,回收率在88.0%~107.4%,RSD为1.7%~9.1%。不同内标物加标量条件下加标回收率和精密度良好,符合GB/T 27404—2008中检测方法确认技术要求。结果表明检验方法准确可靠,满足甲基咪唑检测要求。

表2 甲基咪唑化合物的线性、相关系数、检出限和定量限Table 2 Standard linearity, correlation coefficient, LOD and LOQ of methylimidazole compounds
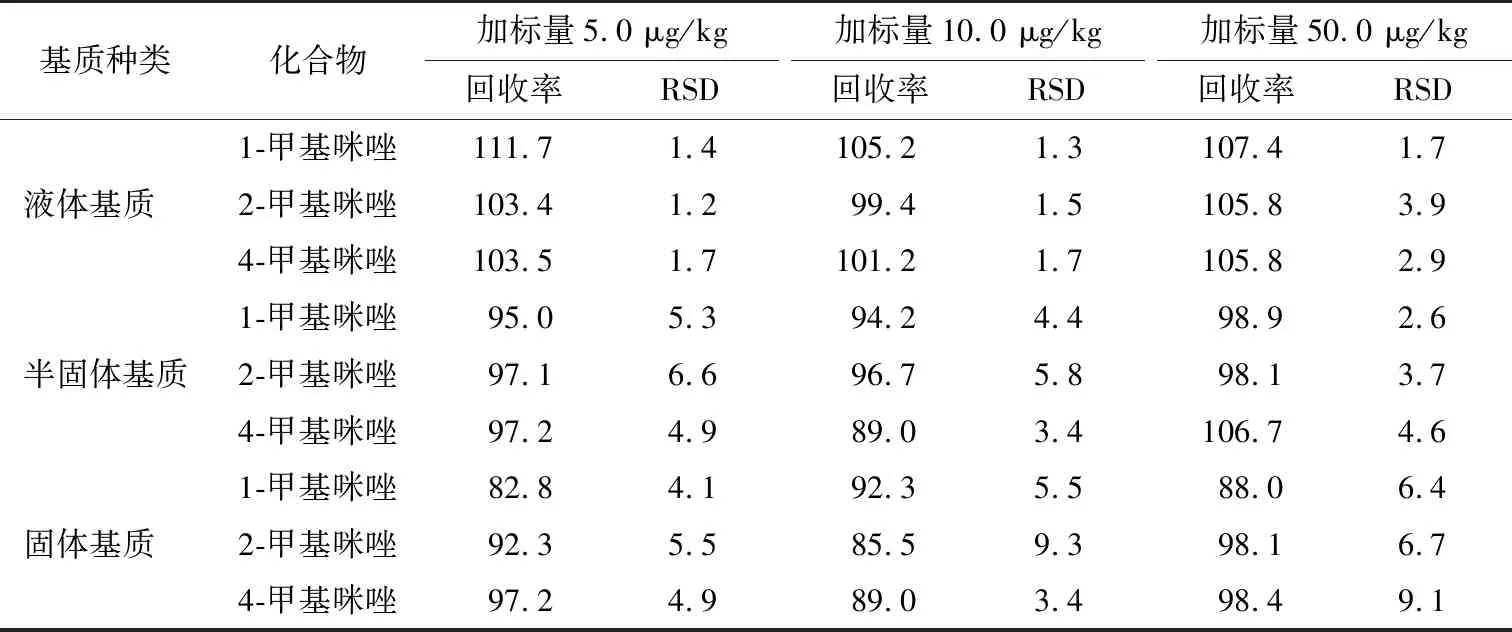
表3 甲基咪唑化合物的回收率和精密度Table 3 Recovery and precision of methylimidazole compounds %
2.7 实际样品检测
采用所建立的方法对市售20种食用香精进行检测,由表4可知,样品中有10份检出上述3种甲基咪唑,检出率为50%。样品中1-甲基咪唑的含量在5.6~32.2 μg/kg,2-甲基咪唑的含量在5.2~160.2 μg/kg,鸡肉粉、牛肉粉和猪骨粉的中2-甲基咪唑含量超过100 μg/kg。4-甲基咪唑的含量在5.1~178.9 μg/kg,牛肉粉和羊肉粉的4-甲基咪唑含量超过了150 μg/kg,其中羊肉粉中4-甲基咪唑含量最高,为178.9 μg/kg,但远低于焦糖中4-甲基咪唑的限量值200 mg/kg。食品中甲基咪唑大多来源于焦糖色素,但市售样品中检出甲基咪唑化合物的香精配料均属肉类香精且均不含焦糖色素,说明肉类香精产品中甲基咪唑化合物来源不是焦糖色素而是肉类酶解物、还原糖和氨基酸发生复杂的美拉德反应而产生的。因此通过控制美拉德反应条件可以控制部分香精产品中甲基咪唑化合物的含量,使产品安全性可控。

表4 市售香精中3种甲基咪唑类化合物的含量†Table 4 Contents of 3 methymidazole compounds in food flavors sold in market μg/kg
3 结论
3种甲基咪唑为同分异构体,分子量小,此类化合物易产生基质效应,因此采用水性柱分离,固相萃取柱吸附净化。采用液相色谱与质谱联用法进行定性分析,使用内标法定量,降低了基质效应影响,保证了检验方法的可靠性和稳定性。利用建立的方法验证了食用香精中甲基咪唑化合物检测的适用性,结果显示该方法定量限优于国家标准。而且该方法快速、高效、灵敏度高,可应用于食用香精中3种甲基咪唑的定量检测。后续将对其他类型食品中甲基咪唑含量测定进行方法适用性研究。
猜你喜欢
云南化工(2022年4期)2022-05-17
口腔护理用品工业(2021年4期)2021-11-02
食品安全导刊(2020年18期)2020-12-03
中国科技纵横(2019年23期)2019-02-14
东方企业家(2018年4期)2018-04-19
小资CHIC!ELEGANCE(2017年19期)2018-03-06
海峡科技与产业(2017年1期)2017-03-04
科技视界(2016年26期)2016-12-17
食品工业科技(2014年6期)2014-05-10
中国信息化·学术版(2013年3期)2013-06-25
