
肥胖诱导的骨骼肌萎缩机制研究进展
2022-11-30 07:04郭婧雅张萍赵雨菡李梦杰黄昆仑仝涛
生物技术进展 2022年6期
郭婧雅,张萍,赵雨菡,李梦杰,黄昆仑,2,3*,仝涛,2,3*
1.中国农业大学食品科学与营养工程学院,精准营养与食品质量重点实验室,教育部功能乳品重点实验室,北京 100083;
2.农业农村部转基因生物安全评价重点实验室(食品安全),北京 100083;
3.食品质量与安全北京实验室,北京100083
骨骼肌是人体氨基酸和蛋白质的主要贮存、代谢场所,约占人体总重的40%[1],其质量和功能的完整性对于维持肌肉骨骼系统的正常功能和代谢稳态至关重要[2]。骨骼肌的质量主要取决于蛋白质合成/分解代谢之间的平衡[3]:当合成/分解代谢稳态失衡时,即分解代谢增强或合成代谢减弱,机体蛋白质丢失表现为骨骼肌质量开始下降,骨骼肌质量进一步下降则可能导致骨骼肌萎缩[4]。骨骼肌萎缩的其他症状还包括肌纤维收缩和肌肉力量下降等[5],其中,肌纤维直径的减少或丧失是骨骼肌萎缩最显著的组织病理学特征[6]。
骨骼肌萎缩是一种世界性的慢性进行性肌肉疾病。根据患病方式不同,骨骼肌萎缩可分为原发型肌萎缩和继发型肌萎缩:前者主要是由于先天性或遗传因素导致;后者则是由于骨骼肌的生长、代谢过程受机体内多种病理因素如癌症恶病质[7]、糖尿病[8]、慢性肾病[9]和肥胖[10]等刺激导致[11]。近年来,肥胖及其相关疾病已经迅速成为世界公共卫生的严峻挑战之一。肥胖是机体能量摄入和能量消耗慢性不平衡导致的代谢型疾病,表现为超重、体内脂肪过度沉积等[12]。近年来,中国肥胖人群数量居高不下,2020年一项覆盖中国31个省的横断面研究表明,约33.3%的成年(≥18岁)中国人处于肥胖状态[13]。此外,肥胖状况也日趋低龄化,《中国居民营养与慢性病状况报告》显示6~17岁的儿童青少年超重肥胖率接近20%,6岁以下的儿童达到10%。肥胖人群特别是儿童期肥胖数量的激增,将直接增加成年期的慢性疾病发病风险[14]。研究表明肥胖和饱和脂肪酸是诱导骨骼肌萎缩发展的独立危险因素[15]。在肥胖的发展进程中,由于营养过剩和全身炎症反应易引发骨骼肌萎缩;另一方面,由于骨骼肌萎缩,患者运动机能下降,进一步加重了肥胖症状,从而形成代谢紊乱的恶性循环,严重阻碍了机体的正常生理进程。
骨骼肌萎缩患者肌肉力量下降,机体运动能力受限,身体机能恢复能力受损,给患者和社会造成严重的经济负担。当前针对肥胖诱导的骨骼肌萎缩的治疗策略包括体育锻炼、营养补充、药物治疗等。然而,这些方法只能一定程度缓解病症。目前为止,仍缺乏针对防治肥胖诱导骨骼肌萎缩的安全有效措施。但部分食品/天然产物中存在的植物化学物如葛根素[16]和芹菜素[17]等已被证明能够缓解肥胖诱导的肌萎缩症状,且从食品/天然产物中分离的植物化学物具有来源广、种类多、安全性高等诸多优势。然而,由于这些物质的作用靶点尚不清晰,限制了其进一步的开发、应用,因此,充分了解肥胖诱导的骨骼肌萎缩的发病机制,对寻找缓解/治疗肥胖诱导的肌萎缩靶点和进一步开发利用天然植物化学物具有重要意义。
1 泛素蛋白酶系统
泛素蛋白酶系统(ubiquitin proteasome system,UPS)是真核生物细胞内主要的蛋白质降解系统。UPS激活过程是一个ATP依赖、多蛋白参与的多步骤反应,通常需要泛素(1种含76个氨基酸的短肽)和3种泛素化酶(泛素活化酶E1、泛素结合酶E2和泛素连接酶E3)的协同作用来实现。蛋白质底物在上述3种泛素化酶的协作下,被泛素分子链(泛素分子相互连接形成的一条线性分子链)共价附着,进一步被机体内的26S蛋白酶体识别和降解,与此同时,泛素被释放并继续循环使用[18]。在一系列酶促级联反应中,泛素连接酶E3参与结合靶蛋白并催化泛素连接底物,这是泛素化过程的限速步骤,因此泛素连接酶E3被认为是泛素化激活反应途径的关键酶[19]。
UPS过度激活是已知的骨骼肌萎缩的重要机制之一。研究表明,肥胖伴随着较高的蛋白泛素化水平。Abrigo等[20]发现高脂喂养C57BL/10小鼠肌肉力量下降,横腹肌肌纤维直径减小,横腹肌内泛素化蛋白水平显著上升。Bollinger等[21]发现在饥饿期间严重肥胖人群(BMI:39.0~57.3)的原代骨骼肌细胞中UPS流增加了4%。进一步表明肥胖人群中骨骼肌萎缩信号增强可能与UPS过度激活有关。同时,该研究显示肥胖人群骨骼肌细胞中蛋白质降解能力提高与蛋白酶体活性和泛素连接酶E3活性增强有关,而与蛋白酶体含量变化无关。因此,靶向降低UPS系统蛋白酶体活性而非含量,可能是合成治疗肌萎缩药物的方向之一。
2 自噬溶酶体系统
自噬溶酶体系统是一种广泛存在于真核细胞内的蛋白降解途径之一[22]。自噬溶酶体系统包括巨自噬、微自噬和分子伴侣介导的自噬,其中巨自噬被认为是自噬介导肌肉萎缩的重要组分之一[6]。巨自噬是通过自噬泡来包裹部分胞质和细胞内需降解的蛋白质等形成自噬体,自噬体可与溶酶体融合形成自噬溶酶体;自噬溶酶体可降解其所包裹的内容物,以实现细胞稳态[23]。
研究普遍认为,自噬稳态失调与骨骼肌细胞内蛋白质合成/分解代谢稳态存在着密切的关联:自噬过度激活会加重分解代谢状态下骨骼肌质量损失[24],引起线粒体碎片化并促进肌肉萎缩[25];自噬不足则导致细胞器或错误折叠的蛋白质低效去除,诱发肌萎缩[26]。但关于肥胖诱导的肌萎缩与自噬的关联存在一定争议。
近年来有证据表明,在肥胖状态下,保持适度自噬水平是维持骨骼肌稳态的基础。Fan等[27]提出高脂饮食诱导的SD(sprague dawley)肥胖大鼠在接受50 mg·kg-1CQ(chloroquine,自噬抑制剂)两周后,腓肠肌中肌肉力量(握力)、质量和肌纤维长度等相关参数均显著降低;此外,体外试验也显示CQ作用加剧了棕榈酸诱导的C2C12成肌细胞肌管萎缩,且雷帕霉素(自噬激动剂)处理可逆转这一现象。这说明一定的自噬激活在体内和体外试验中均能对肥胖引起的肌肉萎缩具有保护作用。
研究表明在高脂饮食诱导的SD大鼠比目鱼肌中,骨骼肌萎缩与自噬系统的关联也存在差异。Cho等[28]表明该模型中自噬相关蛋白如Beclin-1蛋白、p62蛋白、微管相关蛋白轻链3和溶酶体相关膜蛋白2等的含量与对照组相比均无显著差异,认为肥胖不影响SD大鼠比目鱼肌的自噬水平。Campbell等[29]发现自噬相关蛋白如Beclin-1蛋白、Atg7蛋白含量没有变化,但Atg12-5蛋白和p62蛋白水平却显著增加。该结果说明不同的自噬相关蛋白表达水平也存在差异,提示肥胖对SD大鼠比目鱼肌自噬系统具有一定影响,这种影响可能指向一个自噬能力增加但自噬流不变的系统,即某些参与自噬的相关蛋白水平增加,但这种水平没有达到自噬流变化的程度[30]。
上述针对SD肥胖大鼠骨骼肌的实验结果表明:肥胖状态下,不同肌肉组织类型中(腓肠肌和比目鱼肌)的自噬作用是存在差异的,这可能与肌肉中肌纤维的种类不同有关;同一组织类型如比目鱼肌中,自噬作用也有差异,考虑到自噬是一个高度的动态过程[31],这种矛盾一定程度上可能与自噬检测方法存在差异有关。
3 胰岛素/IGF1-PI3K-Akt
肌萎缩过程中胰岛素/胰岛素样生长因子1(insulin like growth factor 1,IGF1)-磷脂酰肌醇3激酶(phosphoinositide 3-kinase,PI3K)-蛋白激酶B(protein kinase B,PKB/Akt)信号传导的关键枢纽是Akt,Akt可激活雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR),刺激蛋白质合成;或抑制叉头盒转录因子O(forkhead box O,FoxO)家族来阻止蛋白水解。通过干扰上述过程可影响蛋白质合成/分解稳态,导致骨骼肌质量下降并进一步引起骨骼肌萎缩。
3.1 mTOR
mTOR是哺乳动物雷帕霉素的靶蛋白,是一种进化上保守的蛋白质,属于丝氨酸/苏氨酸激酶[32]。mTOR可参与p70核糖体S6蛋白激酶(p70 ribosomal S6 protein kinase,p70S6K)和4E结合蛋白1(4E binding protein 1,4E-BP1)调节蛋白质合成的过程。mTOR参与激活p70S6K使S6蛋白磷酸化,激活5-TOPmRNA蛋白翻译起始;4E-BP1在被mTOR磷酸化后,释放被抑制的真核生物翻译起始因子4E,激活蛋白翻译起始[33]。mTOR以平行、独立的方式调节p70S6K和4E-BP1的磷酸化过程,促进机体蛋白合成[34]。此外,近年来的研究表明,mTOR也能通过激活翻译起始因子2B来增强蛋白质合成[35]。因此,抑制mTOR活性引起骨骼肌蛋白质合成受阻是诱发骨骼肌萎缩的重要机制之一。
在蛋白合成代谢调节过程中,mTOR能感受细胞环境和上游信号分子的变化并做出响应,其中经典的mTOR激活途径为胰岛素/IGF1-PI3KAkt。研究发现,高脂喂养C57BL/6J小鼠股直肌中葡萄糖转运子4水平、IGF1受体磷酸化水平、Akt磷酸化水平和p70S6K磷酸化水平均显著降低,表明高脂饮食喂养下胰岛素/IGF1-Akt信号通路被抑制,导致下游p70S6K参与的蛋白质合成减少,诱导骨骼肌萎缩发生,该过程很可能与mTOR密 切 相 关[36]。Lee等[37]研 究 表 明 高 脂 喂 养 下C57BL/6N小鼠肌肉质量、肌肉力量(握力)均显著下降,腓肠肌中PI3K磷酸化水平、Akt磷酸化水平和mTOR磷酸化水平均下降,蛋白合成代谢受阻;而接受4周全麦谷物补充后(补充前后饲料总能量和脂肪组成比例保持一致)可激活高脂诱导肥胖小鼠体内的PI3K-Akt途径,上调mTOR磷酸化水平,改善肥胖引起的肌肉萎缩。除了胰岛素/IGF1-PI3K-Akt激活途径,mTOR也可以被其他物质直接激活,如氨基酸已被证明可通过抑制TSC复合体亚基1/TSC复合体亚基2或通过激活Ras相关小分子G蛋白Rheb来上调mTOR[38],磷脂酸可通过直接结合mTOR来激活蛋白质合成信号[39]。因此,调控胰岛素信号通路或下游mTOR是探索缓解肥胖诱导骨骼肌萎缩的方向之一。
3.2 FoxO
叉头盒(forkhead box,Fox)家族是一类DNA结合区具有翼状螺旋结构的转录因子,根据DNA结合区的同源性,已在不同种属中证实了100多种Fox家族成员,分属A~Q共17个亚族,其中FoxO转录因子是肌萎缩过程中分解代谢反应的关键介质。FoxO家族相关转录因子的磷酸化过程被抑制后,导致FoxO转录因子易位到细胞核,促进肌萎缩相关基因表达上升,如FoxO1可增加两种肌萎缩相关基因即肌肉萎缩盒F基因(muscle atrophy F-box protein,MAFbx/atrogin-1)和肌肉环状指基因1(muscle-specific ring finger protein 1,MuRF-1)转录激活,进而导致肌萎缩发展[40]。Cheng等[41]发现高脂喂养C57BL/6J小鼠的腓肠肌表现出PI3K水平、Akt磷酸化水平和FoxO1磷酸化水平显著下降,同时FoxO1水平显著上升,认为在肥胖小鼠骨骼肌中可通过抑制Akt信号传导来促进FoxO1激活,进一步导致骨骼肌萎缩;而紫草酸镁B可抑制FoxO1表达以缓解肌萎缩。此外,一项研究表明FoxO3转录上调也可能与肌萎缩相关,高脂喂养SD大鼠比目鱼肌中FoxO3水平上调,atrogin-1/MAFbx转录活性增强,进一步导致肌萎缩[42]。但在肥胖状态下,上述FoxO1和FoxO3是否具有协同作用,以及其他FoxO转录家族成员如FoxO4是否也积极参与这一过程尚不清楚,仍需进一步探索。
综上所述,胰岛素/IGF1-PI3K-Akt信号通路下游包括mTOR和FoxO家族,前者可通过激活p70S6K和4E-BP1来刺激蛋白质合成,后者可通过增加MAFbx/atrogin-1和MuRF-1转录激活来诱导蛋白水解。因此,精细化调控胰岛素/IGF1-PI3K-Akt信号通路响应过程,可能是探索缓解肥胖诱导的骨骼肌萎缩的途径之一。
4 肌肉生长抑制素
肌肉生长抑制素(myostatin)是转化生长因子-β超家族(transforming growth factor-β,TGF-β)的一员,也称为生长分化因子8。它是一种由肌细胞产生和释放的蛋白质,特异表达于胚胎期以及成体的骨骼肌,是骨骼肌生长的有效负调节剂[43]。肌肉生长抑制素可使Ⅱ型丝氨酸/苏氨酸激酶受体ActRⅡB活化并与其结合,激活活化素受体样激酶4和活化素受体样激酶5,导致Smad2/3的磷酸化[44]。活化的Smad蛋白在肌生长抑制素易位到细胞核时作为肌肉生长抑制素信号传导的关键细胞内介质,并通过与DNA和其他核因子的相互作用激活靶基因的转录,导致蛋白质合成减少进而引起骨骼肌萎缩[45]。此外,一种线粒体衍生肽MOTS-c被认为能通过减少肌肉生长抑制素的表达来减少高脂饮食诱导的肌肉萎缩信号传导[46]。
高脂喂养下C57BL/6小鼠腓肠肌中肌肉生长抑制素mRNA水平、Smad2和Smad3磷酸化水平均显著上升,且肌肉质量下降,但通过肌肉生长抑制素抑制剂干预后上述现象均有显著改善[47]。此外,在28名严重肥胖人体内,Amord等[48]发现血清中肌肉生长抑制素水平显著升高,且与HOMA稳态模型中胰岛素抵抗指数呈正相关,推测肌肉生长抑制素与胰岛素抵抗存在相关性,并建议进一步评估其作为治疗肥胖诱导的肌萎缩等代谢并发症的药理学靶点。Tanaka等[49]认为在肥胖性高胰岛素血症患者中,肌肉生长抑制素对骨骼肌生长的抑制作用更高,并形成恶性循环,加剧了肥胖患者的肌肉萎缩和能量代谢紊乱。综上所述,肌肉生长抑制素作为骨骼肌质量的关键调节因子,不仅可以单独导致蛋白质合成减少进而引起骨骼肌萎缩,而且在肥胖状态下,可能与其他肌萎缩诱导因子之间具有复杂的协同效果。
5 白细胞介素-6
白细胞介素-6(interleukin-6,IL-6)简称白介素6,是最重要的促炎因子之一。早期研究中IL-6已被认为与骨骼肌萎缩有关,在大鼠或小鼠中高剂量或长期施用IL-6会导致骨骼肌中蛋白质降解增加[50]。此外,IL-6也被报道可对IGF1轴产生干扰,如转基因IL-6小鼠模型中观察到IGF1水平显著降低,细胞因子信号转导抑制蛋白3的mRNA表达增强[51],细胞因子信号转导抑制蛋白3可促进UPS系统激活增加蛋白分解效应[52]。
近年来,研究提出IL-6也可能通过Janus激酶/信号转导与转录激活子3信号通路(Janus kinase/signal transducer and activator of tran-scriptions 3,JAK/STAT3)发挥作用,即IL-6与IL-6R结合形成IL-6/IL-6R/gp130复合物,激活JAK蛋白,磷酸化转录因子STAT3,STAT3易位到细胞核中影响肌萎缩相关基因的转录[53]。Pellegrinelli等[54]发现内脏脂肪细胞与人成肌细胞3D共培养后环境中促炎水平增强,其中IL-6含量显著上升,同时高脂饮食诱导的肥胖小鼠中也发现了该现象,提示促炎因子IL-6可能是肥胖诱导的肌萎缩驱动因素。Andrich等[55-56]发现对Wistar大鼠进行14 d高脂喂养后,比目鱼肌比力(每单位肌肉组织产生的力)明显下降,之后对比目鱼肌中IL-1B、IL-18、IL-33和IL-6含量进行测定,结果显示只有IL-6水平显著上升,而其他白细胞介素无显著变化。上述研究结果表明在肥胖状态下,IL-6相比其他白细胞介素可能具有特异的促肌萎缩作用。
6 肿瘤坏死因子
肿瘤坏死因子(tumor necrosis factor,TNF-α)是一种主要由巨噬细胞和单核细胞分泌的促炎细胞因子。肥胖状态下脂肪组织产生TNF-α,其被转运到骨骼肌并激活核转录因子kappa B(nuclear factor kappa B,NF-κB)信号传导,正常情况下,NF-κB和核因子κB抑制蛋白(inhibitor of NF-κB,IκB)形成复合物存在于细胞质中,但当TNF-α被转运且诱导IκB激酶(IκB kinase,IKK)活化后,IKK可触发IκB的磷酸化,NF-κB被释放易位到细胞核中刺激萎缩相关基因(如MAFbx/MuRF-1)的表达,触发UPS系统导致骨骼肌退化[57]。此外,TNF-α也能通过c-Jun氨基末端激酶信号途径,抑制胰岛素受体激活,是胰岛素/IGF1-Akt途径的潜在抑制剂[10]。研究表明TNF-α可激活NF-κB和c-Jun氨基末端激酶1,诱导C2C12细胞中肌萎缩相关基因的表达和胰岛素抵抗[58]。此外,高脂喂养C57BL/6J小鼠骨骼肌中IκB、NF-κB磷酸化水平上升,可能是骨骼肌中饱和脂肪酸激活炎症小体NLRP3后加剧了上述磷酸化过程,促进肌萎缩发生[59]。综上所述,在肥胖状态下,机体产生的TNF-α可能通过激活UPS或抑制胰岛素/IGF1-Akt等多种途径诱导骨骼肌萎缩发生。肥胖诱导的骨骼肌萎缩过程涉及的信号分子通路见表1。
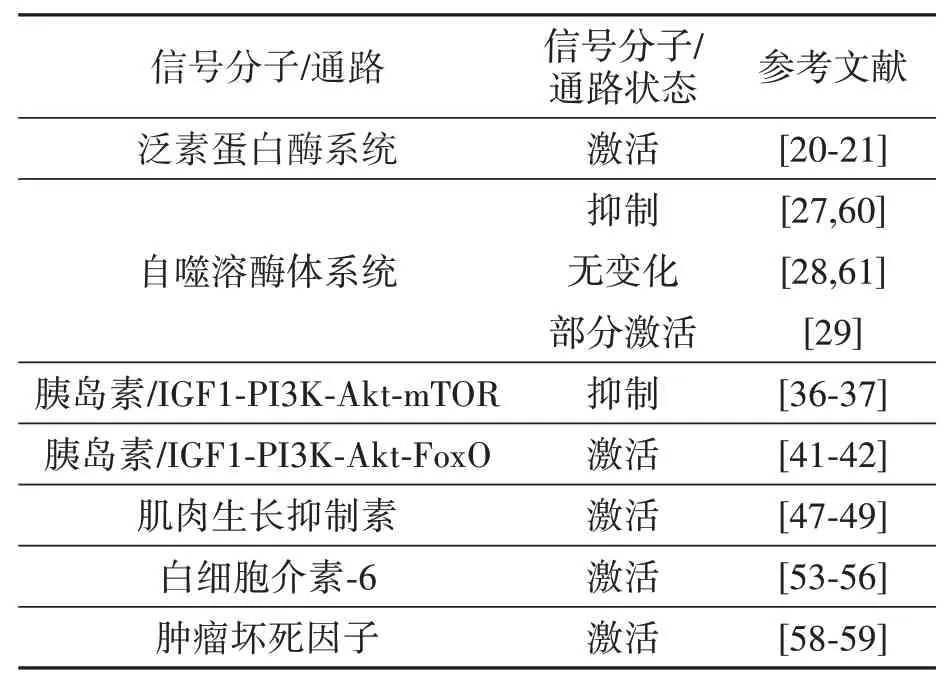
表1 肥胖诱导的骨骼肌萎缩过程涉及的信号分子/通路Table 1 Signaling molecules/pathways involved in obesityinduced skeletal muscle atrophy
7 异位脂质沉积
正常骨骼肌中只有少量脂滴存在,而肥胖状态下由于游离脂肪酸产生过多,超过了组织的氧化能力,造成骨骼肌等非脂肪组织发生脂质堆积,即异位脂质沉积。骨骼肌中脂肪沉积部位主要发生在骨骼肌纤维间和骨骼肌细胞内[62]。研究表明高脂饮食喂养下C57BL/6J小鼠股直肌肌肉质量、与肌萎缩相关基因atrogin-1/MAFbx和MuRF-1表达显著下降,油红O染色显示其骨骼肌出现明显异位脂质沉积;此外,磷酸化蛋白质组学分析显示高脂喂养小鼠的骨骼肌中与炎症相关的磷酸化蛋白(包括血管紧张素转化酶、丝氨酸/苏氨酸蛋白激酶RIO3等)显著表达[36]。高脂喂养巴马迷你猪也具有异位脂质沉积特征,表现为大量脂肪储存于骨骼肌中;且肌肉质量、肌纤维面积(背长肌、股四头肌、腓肠肌和比目鱼肌)显著下降,其中脂肪组织分泌的11β-羟色胺脱氢酶1型可将皮质酮转化为皮质醇,皮质醇能够促进骨骼肌中肌生成抑制素合成,进而促进肌萎缩发展;此外,通过DNA微阵列测定背长肌基因表达谱后,结果表明差异表达基因主要集中在炎症、氧化应激等[63]方面。Verpoorten等[64]发现脂肪酸转运蛋白CD36缺陷小鼠在高脂饮食状态下,也伴随骨骼肌(股四头肌)异位脂质沉积,认为骨骼肌异位脂质沉积浸润可能导致卫星细胞活化能力受损,骨骼肌再生能力减弱,进一步引起骨骼肌萎缩,且提出CD36是异位脂质沉积引起肌萎缩的关键靶点之一。截至目前,异位脂质沉积引起肌萎缩机制尚不明晰,但可能与脂质和骨骼肌存在串扰(crosstalk)有关:脂质能够以自分泌或旁分泌的方式分泌各种脂肪因子,它们不仅能够引起炎症,而且可能通过影响卫星细胞干扰骨骼肌稳态。
8 线粒体功能障碍
近年来,研究表明线粒体功能障碍与骨骼肌萎缩存在关联。高脂喂养C57BL/6小鼠表现出骨骼肌萎缩症状,主要为腓肠肌中线粒体呼吸链复合体Ⅰ/Ⅱ水平、柠檬酸合酶活性和氧化磷酸化相关蛋白mRNA水平显著下降,说明线粒体电子传递链活性和氧化能力均降低。此外,高脂喂养也影响了线粒体生物发生,例如高脂喂养C57BL/6小鼠腓肠肌中线粒体形态较小且聚集程度下降,同时线粒体生物发生相关基因如过氧化物酶体增殖活化受体γ辅助活化因子1α、线粒体转录因子A和细胞色素C水平显著下降[17]。有研究者认为这种关联可能与线粒体内过量活性氧的产生有关,活性氧水平过高可导致与骨骼肌功能受损相关的氧化应激水平升高,加速肌肉质量损失[65]。此外,也有人认为这与线粒体可能在触发分解代谢信号方面有重要作用有关[66-67]。因此,仍需更多研究来探究线粒体功能障碍与肥胖诱导的骨骼肌萎缩间的关联。
9 展望
随着全球肥胖问题日趋严重,肥胖诱导骨骼肌萎缩的发病率也逐年攀升,已成为公共卫生安全的重要挑战之一。一方面,由于目前缺少缓解/治疗该疾病的高效靶向药物,因此充分了解其发病机制刻不容缓。另一方面,食品/天然产物中存在的植物化学物不仅具有来源广泛、种类繁多、安全性高等诸多优势,且部分物质已被证明具有缓解肥胖诱导的肌萎缩症状的潜在效力,但仍存在作用靶点不清晰等问题。因此,本文系统总结和讨论了肥胖诱导骨骼肌萎缩的主要影响机制,包括泛素蛋白酶系统、自噬溶酶体系统和胰岛素/IGF1-PI3K-Akt信号通路等。上述诸多影响机制除了可单独发挥作用外,也呈网络状相互交错、互相影响,共同调节骨骼肌蛋白质的合成/分解稳态。因此,在已有研究基础上,应更注重探索骨骼肌蛋白质合成/分解调控的精细化机制。同时,应积极开发食品/天然产物的候选化合物研究,验证有效性并评价其安全性,并推动临床应用及产业化。
猜你喜欢
中华养生保健(2020年5期)2020-11-16
医学综述(2020年11期)2020-02-16
中国运动医学杂志(2016年3期)2016-07-10
现代电生理学杂志(2016年1期)2016-07-10
中西医结合心脑血管病杂志(2016年20期)2016-03-01
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
医学研究杂志(2015年12期)2015-06-10
中国康复理论与实践(2015年7期)2015-05-09
中国医学科学院学报(2015年5期)2015-03-01
西安交通大学学报(医学版)(2015年2期)2015-02-28
