
联合TCGA和GEO数据库构建由circRNA介导的非小细胞肺癌特异性竞争性内源性RNA网络
2023-06-07 05:41刘金婵许德华李让唐田书陈铭陈晓琳饶绍奇
河北医药 2023年7期
刘金婵 许德华 李让 唐田书 陈铭 陈晓琳 饶绍奇
在2020年全球癌症统计中,肺癌是第二常见,也是死亡率最高的癌症,对人类健康构成了严重的威胁。肺癌分为小细胞肺癌(small cell lung cancer,SCLC,15%)和非小细胞肺癌(non-small cell lung cancer,NSCLC,85%)[1];NSCLC早期患者以手术治疗为主,Ⅲ期是同步放化疗后免疫治疗,但患者存活率均较低[2]。因此,找到与NSCLC发生发展相关的分子机制,发现有效的早期诊断生物标志物以及临床治疗的潜在靶点,改善患者的生存和预后,极为重要。circRNA存在共价闭合的单链环构象,不易受到破坏,并且存在一个或多个miRNA反应元件(miRNA response element,MRE)[3],可基于竞争性内源性RNA(competing endogenous RNA,ceRNA)机制参与癌症发展过程中,如circRNAs可作为ceRNA,与miRNA中的MRE结合,减少miRNA与其靶mRNA结合,从而调节miRNA对靶mRNA的抑制作用。先前研究认为circRNA与各种疾病的发展过程都具有显著地相关性,尤其是关于癌症的进展过程[4],同时,circRNA在癌症细胞和各种体液[5]中特异表达,反映出circRNA作为NSCLC诊断生物标志物或治疗靶点的极大潜力。在这项研究中,从TCGA数据库和GEO数据库获取的芯片数据,得到NSCLC中差异表达的circRNA、miRNA 和 mRNA并通过这3种RNA来进行ceRNA网络的构建并识别出网络中的核心circRNA,并从中提取了NSCLC特异性ceRNA核心子网络;最后把 ceRNA 机制作为探索非NSCLC发展过程的出发点,来进一步说明核心 circRNA在其中是如何通过调控基因的表达从而作用于癌症的发展机制,为开展NSCLC的致病机制、诊断生物标志物与治疗潜在靶点等研究提供参考。
1 资料与方法
1.1 一般资料 本研究使用的3个circRNA芯片数据GSE101586 (5病例,5对照),GSE101684 (4病例,4对照) 和 GSE112214 (3病例,3对照)均来源于GEO(gene expression omnibus)数据库。494例NSCLC的miRNA-seq、RNA-seq及临床信息则来自TCGA(the cancer genome Atlas)数据库。
1.2 差异表达circRNA (DEcircRNA)的筛选 首先,对GEO数据库的circRNA数据规范化和以及对数据进行log2的变换,之后采用“limma” R软件包进行差异表达分析,以|log2(Foldchange)| >1和P-value<0.05为标准确定每个数据集中差异表达的circRNA (DEcircRNA)。然后,使用“Robust Rank Aggreg”R软件包的稳健的秩集聚法(robust rank aggregation,RRA)根据表达水平和P-value对3个数据集的DEcircRNA进行了整合和排序,确定最优的DEcircRNA。
1.3 差异表达mRNA(DEmiRNA)和miRNA(DEm R
NA)的筛选 首先,对TCGA数据库NSCLC的RNA-seq和miRNA-seq数据过滤,将表达量较低的基因删除(过滤标准为:平均表达值<1);随后,进行差异分析,以FDR校正的P value <0.05和|log2(Foldchange)| >1的标准得到DEmiRNA、DEmRNA的新表达矩阵。
1.4 ceRNA 网络的构建和核心网络的提取 首先,从circbase数据库[6]获取DEcircRNA的fasta序列和从miRbase数据库[7]获取DEmiRNA的fasta序列。随后,利用miRanda[8]、 RNAhybrid算法[9]预测存在结合靶位的circRNA-miRNA调控关系,两个算法均预测到的调控对将纳入网络构建。然后,从miRTarbase数据库[10]中得到DEmiRNA和DEmRNA之间的调控关系。根据对子中相同的 miRNA 进行 ceRNA 网络的构建,最终得到了多条完整的circRNA-miRNA-mRNA调控轴,根据miRanda算法识别出结合位点数>4的circRNA作为核心节点并提取其调节的子网络。最后使用 Cytoscape 软件(版本 3.6.1)可视化。
1.5 功能富集分析和生存分析 首先,利用 “cluster Profile” R包进行GO和KEGG功能通路分析,探讨子网络的生物学功能。然后,将从上述的DEmRNA新表达矩阵和临床信息整合,再使用 “survival” R包对核心子网中的基因进行Kaplan-Meier分析。
2 结果
2.1 筛选差异表达的 RNA circRNA的差异分析结果显示,GSE101586、GSE101684、GSE112214芯片分别得到180个、1 341个、587个DEcircRNA。整合3个数据集结果,经RRA算法确定了42个DEcircRNA。RNA-seq得到5 537个DEmRNA。miRNA-seq得到362个DEmiRNA。
2.2 构建非小细胞肺癌特异性circRNA-miRNA-mRNA网络并提取其核心子网络 基于miRanda、RNAhybrid算法,本研究得到了642个circRNA-miRNA调控对子;然后,从miRTarbase数据库获取了838个miRNA-mRNA调控对子。借助共享的miRNA,构建了包含35个circRNA,77个miRNA,283个mRNA的circRNA-miRNA-mRNA网络。进一步以结合位点数>4的8个circRNA为核心,并以其调控的分子(miRNA和mRNA)为目标节点,提取了1个包含56个节点(8个circRNA,10个miRNA,38个mRNA)、61条边的子网络。见图1。
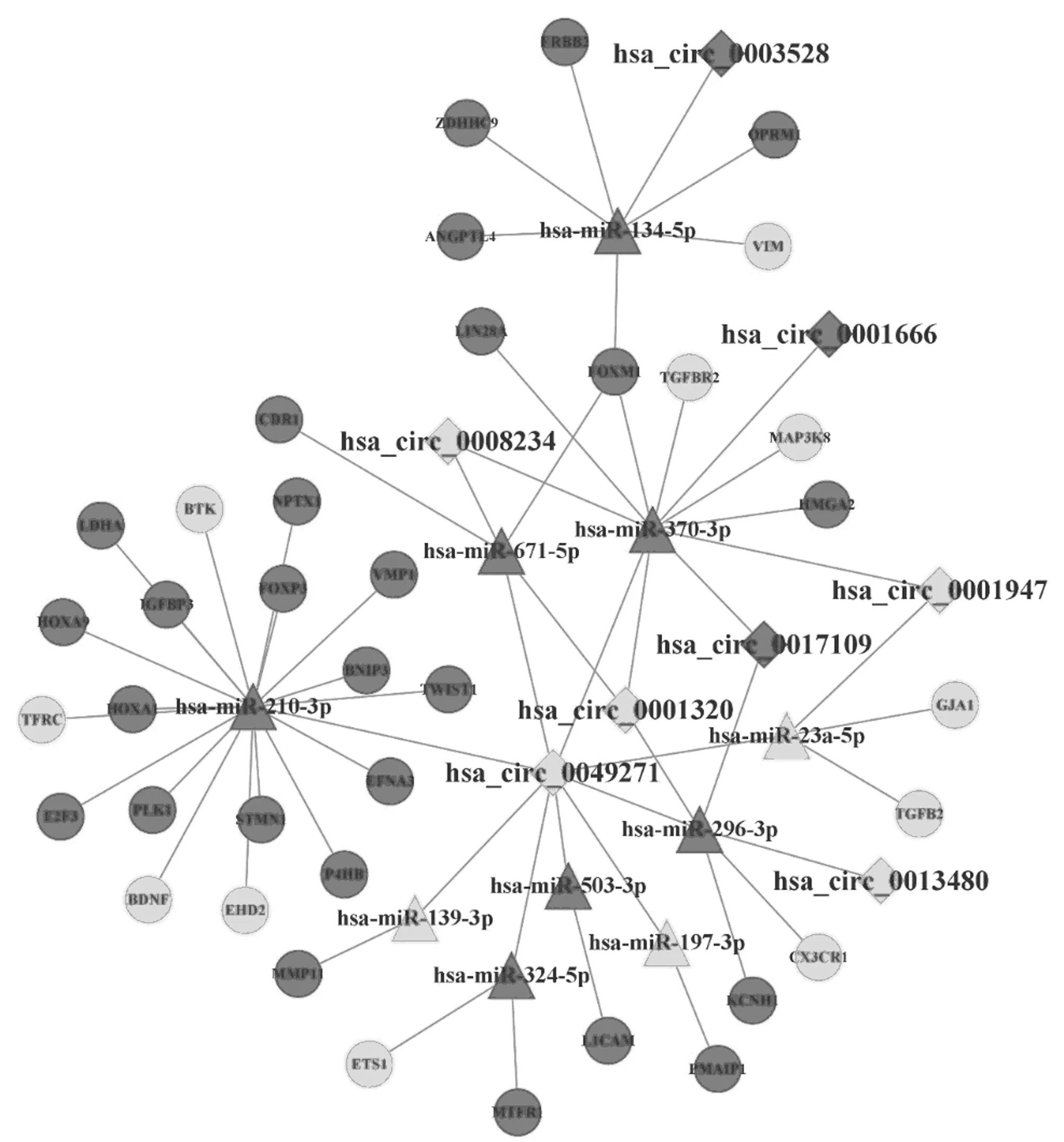
图1 NSCLC特异性ceRNA核心子网络;菱形、三角形和圆形分别代表DEcircRNA、 DEmiRNA 和 DEmRNA
2.3 非小细胞肺癌特异性ceRNA核心子网络的生物
学意义 对GO的富集分析显示,该ceRNA核心子网络、显著富集于由中性粒细胞激活参与的免疫反应、细胞对化学应激的反应、核受体活性,配体激活转录因子活性等功能;对KEGG数据库的富集分析显示,该ceRNA核心子网络主要富集于MAPK信号通路、p53信号通路、非小细胞肺癌等通路。以上结果表明NSCLC特异性ceRNA调控网络涉及多个生物学过程和信号通路。
2.4 非小细胞肺癌特异性ceRNA核心子网络的的临床意义 生存分析表明NSCLC特异性ceRNA核心子网络中有14个基因显著影响患者的预后情况(P<0.05)。与低表达组比较,ANGPTL4、FOXM1、HMGA2、HOXA1,OPRM1,PMAIP1,LDHA,TWIST1,MTFR1,PLK1基因高表达组的预后较差;而MAP3K8、TGFBR2、BTK、CX3CR1基因高表达组的预后较好(或解释为保护因子)。见图2。
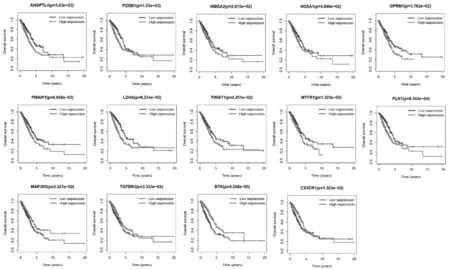
图2 NSCLC特异性ceRNA核心子网络中14个预后基因标记的生存曲线
3 讨论
circRNA作为一种新的诊断标志物和治疗分子,已被研究并应用于多种疾病的诊断和治疗中。目前多个研究均基于单个cirRNA,而复杂疾病的发展是由于多个生物分子相互作用的复合物的扰动引起的[11]。此外,在ceRNA网络中,一个circRNA竞争性吸附多个miRNA,一个miRNA也可被多个circRNA吸附,导致下游基因的表达异常。基于以上事实,本研究希望通过构建包含circRNA、miRNA和mRNA的完整ceRNA网络,系统地阐明NSCLC的病理机制并识别与其预后相关的分子标记。然而在NSCLC的以往研究中,较少有针对circRNA介导的ceRNA调控网络的全基因组研究。本研究通过充分结合两个数据库,构建了由circRNA介导的NSCLC特异性ceRNA调控网络,从中提取出连通度较高的节点以及由节点所介导的核心子网络。最后,通过富集分析和生存分析,阐明了NSCLC特异性ceRNA核心子网络的生物学意义和临床意义。
ceRNAs和miRNAs共享MRE的数量被证明是ceRNA交叉调节的关键决定因素[12],具有异常高密度结合位点的circRNA可能会在疾病网络中发挥着关键作用。本研究利用miRanda算法挖掘到8个核心的circRNA,其中hsa_circ_0001666/0008234/0001947均已被发现与肺癌相关。hsa_circ_0001666高表达与NSCLC 患者淋巴结转移和病理分期呈正相关[13]。而hsa_circ_0001947则在NSCLC组织和细胞中低表达,敲低后增强了细胞活力和增殖能力,诱导细胞生长时停留在S期,最终导致增殖异常[14]。
8个核心circRNA调控的10个miRNA中,hsa-miR-671-5p、hsa-miR-503-3p、hsa-miR-210-3p、hsa-miR-296-3p、has-miR-197已被报道与NSCLC密切相关,如miR-671-3p在NSCLC组织和细胞系中表达上调[15],通过调节FOXP2促进癌细胞增殖、凋亡和迁移。hsa-miR-503-3p在NSCLC中表达上调,并发现其在癌症原发性耐药中发挥重要作用[16]。其余5个miRNA虽暂时未见有报道与NSCLC相关,但均被发现与一种或多种癌症相关。上述说明本文识别的与circRNA有调控关系的10个miRNA在NSCLC或其他癌症中发挥重要作用,其生物学意义是明确。
本研究通过对特异性ceRNA核心子网络进行功能通路富集分析发现,子网络主要参与到中性粒细胞激活参与免疫反应、配体激活的转录因子活性、MAPK信号通路、p53信号通路、非小细胞肺癌等。中性粒细胞是免疫细胞家族的关键成员,激活后表达大量膜受体,识别和消除感染因子[17]。KEGG富集结果中的大部分通路已有文献报道与NSCLC发生机制密切相关,如经典的p53信号通路[18]作为一条抑癌通路广泛参与到NSCLC中。另外,子网还直接富集到NSCLC的通路上。以上信息提示,本研究所筛选的核心cirRNA介导的ceRNA网络密切涉及了NSCLC整个发展过程。
生存分析表明NSCLC特异性核心子网络中有14个基因显著影响患者的预后情况,而网络中的8个核心circRNA很可能通过ceRNA调控网来调节这些基因,影响其表达,对患者预后产生作用。例如:在hsa_circ_0001320/0001947/0008234-hsa-miR-370-3p-MAP3K8/TGFBR2调控轴中,cirRNA下调、miRNA上调、mRNA下调,3个circRNA的低表达会减少与miR-370-3p的结合,使更多的miR-370-3p与MAP3K8、TGFBR2直接结合,导致MAP3K8、TGFBR2在体内的含量降低。TGFBR2的缺失会增加肿瘤侵袭性并降低肺腺癌的存活率[19]。
而本文生存分析的结果以及相关研究[20]都表明,MAP3K8、TGFBR2基因表达量低的患者,生存率相对较低。因此,推测hsa_circ_0001320/0001947/0008234等circRNA的低表达可能与NSCLC患者的不良预后相关。在hsa_circ_0049271-hsa-miR-210-3p-BTK这一条调控关系上,circ_0049271表达下调、miR-197-3p、BTK表达上调,说明circ_0049271下调可导致BTK的低表达,BTK调节p53的活性以增强细胞凋亡、衰老反应以及肿瘤抑制反应[21]。生存分析同时也显示BTK低表达组患者预后较差,提示hsa_circ_0049271低表达与NSCLC患者不良预后相关。总之,本文通过构建NSCLC特异性ceRNA核心子网络识别出来的8个circRNA可能通过与一个或多个miRNA结合,以此来调节与预后显著相关基因的表达,进而影响NSCLC患者总体预后情况。
然而,我们也注意到本研究的不足之处。(1)用于筛选差异表达circRNA的三套数据样本量偏小,可能会缺少足够的统计功效识别微小或中度效应的circRNA。(2)部分circRNA、miRNA和mRNA之间的调控关系是通过计算机算法识别的,缺少实验室的证据。(3)研究所选的数据针对西方群体,因此本次研究是否能推广和应用到中国人群中,仍然需要进行探讨。本研究通过整合多组学的数据,系统性分析了circRNA、miRNA和mRNA之间的调控关系,并构建NSCLC特异性ceRNA核心子网络。进一步功能富集分析和生存分析表明NSCLC特异性ceRNA核心子网络具备清晰的生物学意义和临床意义,在8个circRNA介导的基因中,有14个基因显著影响患者的预后情况。
猜你喜欢
学苑创造·A版(2020年12期)2020-01-07
中国外汇(2019年15期)2019-10-14
中国医疗保险(2017年5期)2017-05-17
作文教学研究(2016年1期)2016-07-05
三峡大学学报(自然科学版)(2016年6期)2016-04-16
中国康复理论与实践(2015年10期)2015-12-24
中国病理生理杂志(2015年8期)2015-12-21
现代电生理学杂志(2015年1期)2015-07-18
医学研究杂志(2015年8期)2015-06-22
医学研究杂志(2015年3期)2015-06-10
