
多相催化CO2 参与的炔烃C-H 键羧基化反应研究进展
2023-10-14 03:34吴洁文付慧宇梁长海
燃料化学学报 2023年9期
吴洁文,付慧宇,陈 霄,梁长海
(大连理工大学化工学院 先进材料与催化工程实验室,辽宁 大连 116024)
工业化进程的加速导致人类活动向大气中大量排放CO2[1,2]。根据国际能源署的最新分析数据,2021 年,全球与能源相关的CO2排放量增加了6%,达到363 亿吨,创造了新的历史记录[3]。CO2作为一种温室气体,其大量排放是导致全球气候变暖的主要原因,与此同时产生了一系列的环境问题[4]。为此,控制CO2在大气中含量成为十年内全球关注的焦点[1,5]。中国政府高度关注全球气候变暖问题,积极履行国际承诺,将2030 年达到碳峰值,2060 年实现碳中和作为国家环境目标。减少大气中CO2含量的两种方法为:CO2的捕捉、封存和CO2的利用[6,7]。碳捕捉和碳封存(CCS)是减少大气中CO2含量最高效的方法[5],而CO2的利用是降低CCS 工艺成本的另一重要因素,同时也是更有效地减少CO2的排放和增加资源利用率的途径[8,9]。CO2的利用可分为直接利用和间接利用,直接利用包括将CO2用于石油开采、焊接保护气、食品添加剂和消防等;间接利用是将CO2进行化学转化制备燃料和化学品等。CO2的转化大致可分为以下几类,催化氢化重整、进行偶联或缩合反应、光电还原和生物转化,由此制得甲醇、有机碳酸酯、氨基甲酸酯、羧基化产物、甲烷和合成气等化学品[10,11]。
CO2的标准吉布斯自由能为-394.38 kJ/mol,分子中的碳原子处于最高氧化价态,整个分子处于最低能量态,化学性质稳定。因此,通过设计合适的反应途径使得CO2参与的化学反应的吉布斯自由能为负,其中,CO2活化是其化学转化的重要前提,目前主要的活化策略有以下四种,第一,使用高能起始原料与CO2反应,如氢气、不饱和化合物、小分子环化物和有机金属;第二,转化的目标产物为能量较低较稳定的物质;第三,除去反应产物中的某一组分以促进平衡的移动;第四,外界输入能量,如光能和电能[12,13]。CO2与端炔羧化生成丙炔酸衍生物的反应引起了研究者的广泛关注。丙炔酸类化合物是一类非常重要的有机中间体[14],可以合成香豆素、黄酮等杂环化合物,也可以通过脱羧交叉偶联反应制备取代炔烃等光电材料(如图1 所示)。
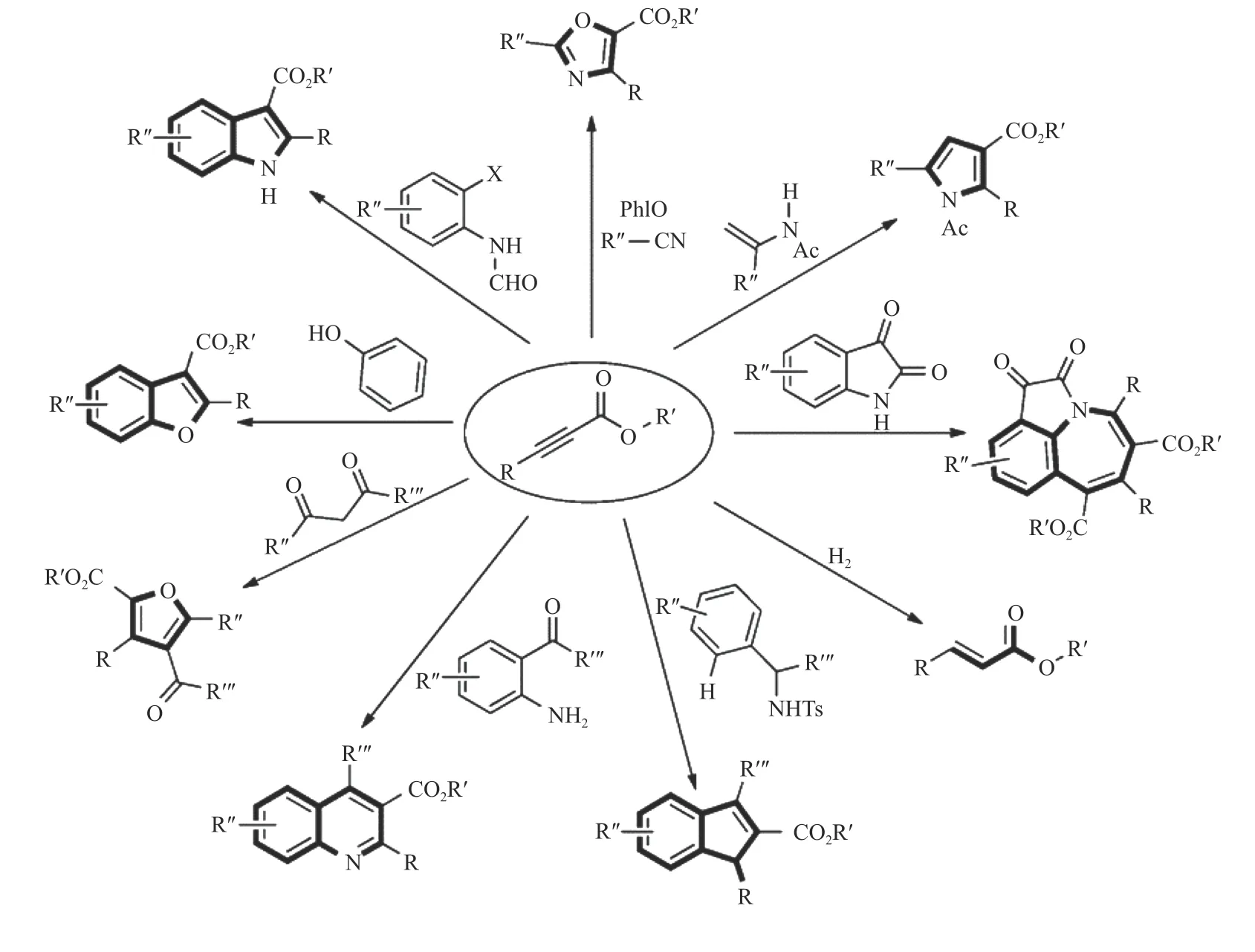
图1 炔酸(酯)类化合物的应用[15]Figure 1 Application of alkynic acid(ester) compounds[15](with permission from American Chemical Society)
关于端炔与CO2羧化生成丙炔酸的催化体系已有较多报道,既有均相反应体系,也有多相反应体系。均相催化系统可以大致分为以下几类,Cu(I)、Ag(I)配合物催化体系;Cu(I)、Ag(I)无机盐催化体系;无过渡金属催化体系和有机催化体系。Cu(I)、Ag(I)配合物催化体系中,{µ-[3,5-(CF3)2Pz]Cu}3化合物可以在无外部碱催化作用下,在室温、大气压力的条件下较好地促进该反应的进行[16]。Yu 等[17]报道了CuCl、配体TMEDA、溶剂DMF 和碳酸钾的催化体系,可以在室温、大气压力下反应16 h达到90%的分离产率。除此之外,还有NHC[17,18]和有机膦配体[19]。在无配体存在下,反应也可以顺利进行。Fukue 等[20]报道了端炔、溴代己烷和K2CO3的混合物在CO2(气球)气氛下,CuI 存在下,在N,N-二甲基乙酰胺中反应4 h,分离得到了炔酸酯。Zhang 等[21]报道了负载量为0.1%的AgI 催化剂体系,该体系与之前报道的10% (IPr)CuCl催化体系的目标产物产率相同都为91%,此外对这两种催化剂系统进行简单的动力学研究表明,AgI/Cs2CO3催化剂系统的活性约为(IPr)CuCl/K2CO3系统的300 倍。Wang 等[22]开发了双组分的无过渡金属催化体系即在低沸点溶剂CH3CN 中,四正丁基醋酸铵和K2CO3存在下,末端炔烃与CO2在相对温和的条件下进行羧化反应。在该体系中四正丁基醋酸铵不仅由于阳离子-Π 键的作用可以活化炔烃,还可以与CO2相互作用,同时它又作为相转移催化剂,增加了碳酸盐在溶剂中的溶解度。有机催化体系中[23],除格林尼亚试剂和有机锂试剂外[24,25],Shi 等[26]报道了一种既含有NHC 又含有羰基的双功能有机催化剂,NHC 可以活化CO2形成NHC-CO2加合物,在Cs2CO3存在下,羰基活化炔烃,且催化剂中羰基与NHC 所形成的钳形结构是影响反应活性的重要因素。虽然均相反应体系的研究取得了一些进展,但是仍存在一些不足,如反应条件苛刻,需要高温、高压;催化剂对水和空气高度敏感;金属负载量高和分离、回收困难等问题[27],体系产生的环境污染和成本效益也不利于进行大规模工业生产[28]。因此,具有易于分离、回收且催化活性高的多相催化体系的设计引起了人们的关注,尤其是在铸币金属基多相催化剂(Ag、Cu 和Au)作用下实现在较温和的反应条件下CO2的高效催化转化。本工作将围绕多相催化体系下炔烃C-H 键与CO2羧基化反应体系的活化、催化剂的结构和反应的机理等研究进展进行系统综述。
1 炔烃C-H 键与CO2 羧基化反应体系的活化
一个反应是否能发生取决其热力学上的数据。经过计算,以端炔和CO2为底物生成丙炔酸的反应路径在热力学上是不能自发进行的(ΔGθ=19.1 kcal/mol)。然而,如果反应是在温和碱的存在下进行,生成羧酸盐,则整个反应在热力学上是可行的(ΔGθ=-10.1 kcal/mol)[29]。在反应可进行的前提下,CO2由于其热力学和动力学上的惰性导致其分子活化是进行化学转化的重要前提。CO2分子呈直线型分子结构,碳原子的杂化方式为sp,电荷排布方式为若想使CO2进行转化,需要克服较高的热力学能垒,通常需要催化剂、高温和高压等条件。CO2的第一电离能(13.79 eV)较大,难以提供电子但具有较低能级的空轨道和较高的电子亲和能(38 eV),相对而言更容易接受电子[30]。因此,活化CO2最有效的手段是采用合适的方式输入电子,即外界提供富电子物质[31-33]与CO2作用,这也是有机合成中最常见活化分子的方法。端炔中三键碳为sp杂化,致使末端炔烃中C-H 键更易于异裂,释放出质子,显示出部分酸性,两个炔碳把Π 电子控制在中心区域,致使三键碳部分裸露[34]。研究证实Cu、Ag 等金属可以与炔烃的Π 电子进行配位达到活化炔烃的作用,而外部N、O 元素的引入也可以与端炔氢形成氢键,进一步极化C-H 键[14,35-38]。反应体系中极性溶剂的存在可以提高碱和CO2的溶解度。因此,通过合理设计反应体系(催化剂、溶剂和碱)可以达到协同活化炔烃C-H 键与CO2的目的。
2 多相催化剂
催化剂的组成和结构变化影响金属和载体的电子结构、化学状态、物理结构、控制反应物的吸附和解吸行为,决定了不同产物的选择性和产率,因此,催化剂的设计和优化是必要的。目前,有三种主要的催化剂设计方法可以提高催化活性:第一种,控制金属活性中心的尺寸和分散程度;第二种,通过控制载体上的电子密度和引入官能团来增强载体对于底物的吸附和活化能力;第三种,设计载体结构降低传质阻力。本工作将系统综述Ag 基催化剂、Cu 基催化剂和Au 基催化剂作用下炔烃C-H 键与CO2羧基化反应。
2.1 Ag 基催化剂
Ag 基催化剂由于其高效的催化性能而被广泛研究。研究者通过对原料组成和制备方法进行巧妙设计,制备出具有高二氧化碳吸附能力、高反应活性和广泛底物适用性的Ag 基催化剂结构,为端炔与二氧化碳的直接羧化反应提供了强有力的支撑,也为后续催化剂的设计提供了思路。Ag 由于独特的d10电子排列使其更容易与C≡C 结合达到活化炔烃的目的,而载体较好的吸附和活化底物的性能使得载体与活性位点之间构成协同催化,保证了高反应活性[35,36]。Shi 等[39]报道的Ag@ZIF-8催化剂(如图2)中Ag 有两种形态:一种为在ZIF-8 表面的纳米Ag 粒子(AgNP);另一种为在ZIF-8 骨架中部分代替Zn2+的Ag+(AgHD)。由于Ag+的化合价较低,但尺寸大于Zn2+,咪唑基向Ag 提供的电子不明显,导致ZIF-8 载体相较于未载Ag 之前具有更高的电子密度。因此,载体与CO2分子中带正电荷的碳原子间相互作用加强,使得载体表面更加富集CO2,而AgNP能够较好地活化炔烃使催化剂整体具有较好的活性(表1,第1 行)。与此类似的Ag/M-CeO2[27]催化剂,介孔CeO2(M-CeO2)具有良好的规整孔道结构,在增加比表面积的同时可以提高反应物质的传质效率。由于载体上氧空位的存在,M-CeO2对CO2具有一定的吸附能力。且Ag 的引入促进了氧空位的形成,增强了载体对二氧化碳的吸附,保证了反应的收率(表3,第1 行)。除以上报道外,MOF 基催化剂中骨架的Lewis 酸碱性质不同直接影响催化剂对底物的吸附能力:具有Lweis 酸性的MOF 骨架对于苯乙炔等炔烃类底物有较强的吸附能力而具有Lweis碱性的骨架对于CO2的吸附能力较强[28,40,41],进而调控反应收率(表1,第2-5 行)。与此类似,Ag@FeNT[42]催化剂由于载体的Lweis 酸性,也具有一定的炔烃吸附能力(表1,第6 行)。
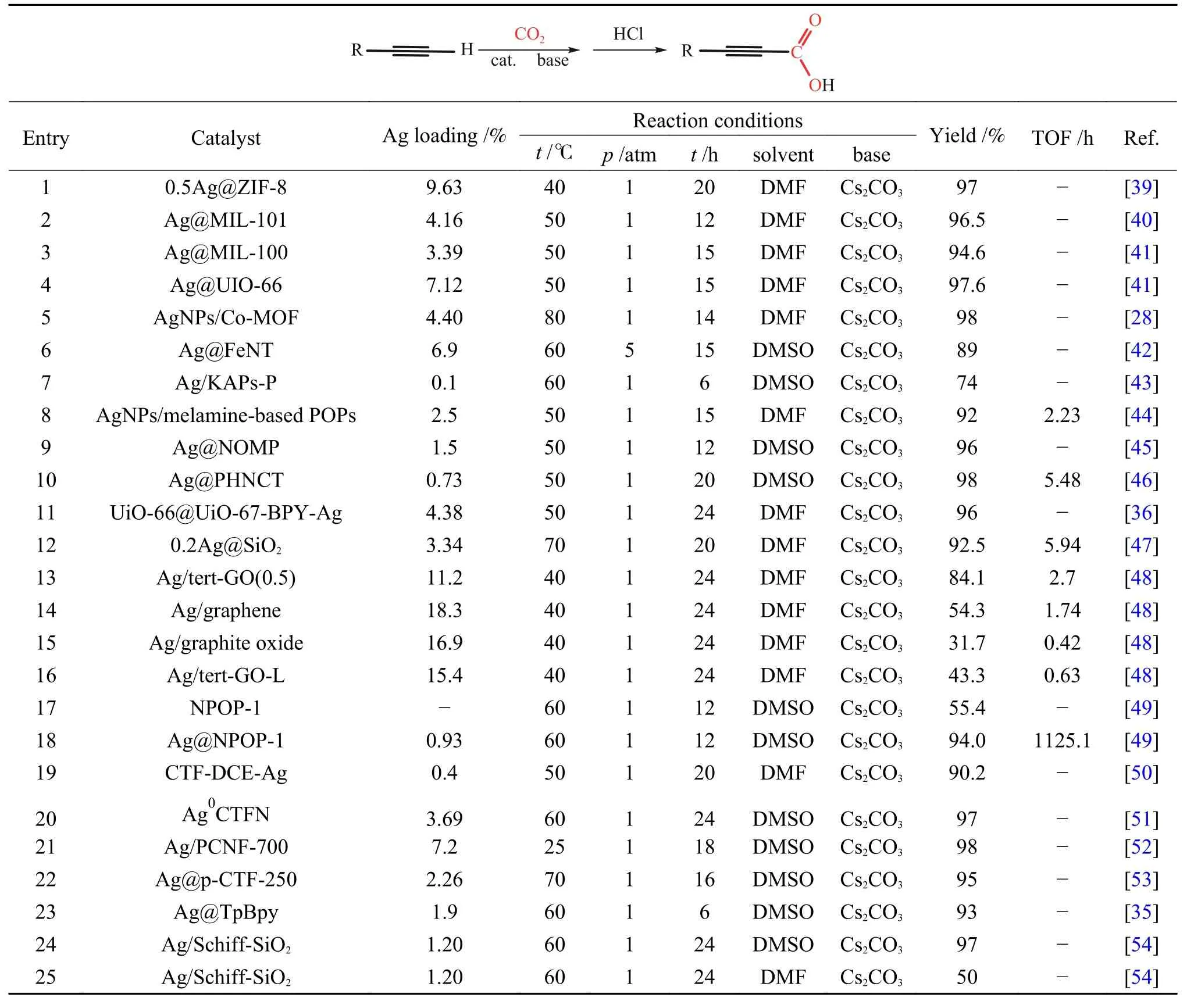
表1 端炔与CO2 直接羧化反应的Ag 基催化体系Table 1 Heterogeneous catalytic system for direct carboxylation of terminal alkynes with carbon dioxide
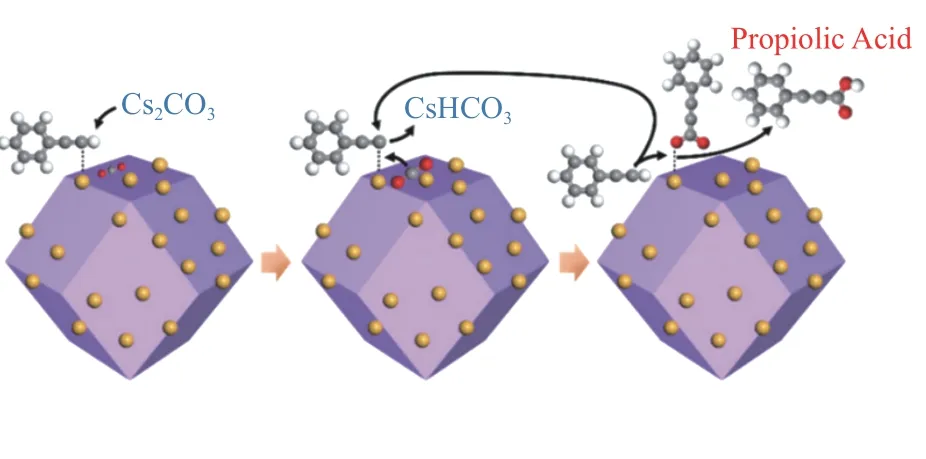
图2 xAg@ZIF-8 CTACO2 反应机理[39]Figure 2 CTACO2 reaction mechanism on xAg@ZIF-8[39](with permission from American Chemical Society)
此外,在催化剂设计中研究人员常会引入一些路易斯碱性位点如氮或磷位点来调控催化剂的性能。如Wu 等[43]通过无水FeCl3催化的Friedel-Crafts反应,将三苯基膦和吡咯为单体编织合成KAPs-P,使用NaBH4还原三氰基甲烷银(AgTCM)获得Ag/KAPs-P 催化剂(如图3(a)所示)。通过HRTEM图像证实,Ag/KAPs-P 中的Ag 纳米颗粒(Ag NPs)均匀分散,平均尺寸为(4.1±1.4) nm 小于对比催化剂Ag/KAPs-Py[(9.8±5.4) nm]和Ag*/KAPs-Py[(17.5±9.6) nm](图3(b)-(c))。在一定的反应条件下(表1,第7 行),当没有催化剂加入时,羧化反应的产率在37%,当加入Ag/KAPs-P 时,产率被提高至74%,而对比催化剂Ag/KAPs-Py 和Ag*/KAPs-Py 的产率分别在66%和59%。此外,无论苯环上连接吸电子基还是给电子基,相应羧化产物的收率可以维持在85%-98%,说明该催化剂对于底物适用性较好。催化剂在最优反应条件下进行了五次循环后,仍能保持苯丙炔酸较高的收率(84%-92%)。同时,五次循环之后Ag NPs 仅有轻微聚集(粒径在(6.0±2.2) nm)。催化剂这一性能上的优越性可归因于磷化氢促进了载体上Ag 组分的分散和稳定:一方面,磷的p电子对可以与Ag 进行配位,有利于将Ag前体分散到KAPs 的介孔中;另一方面,磷的p电子对也有利于Ag 纳米颗粒的锚定,阻碍了其在还原过程中的聚集。因此,可以在KAPs-P 材料上容易地获得小尺寸、稳定性好和分散均匀的Ag NPs。
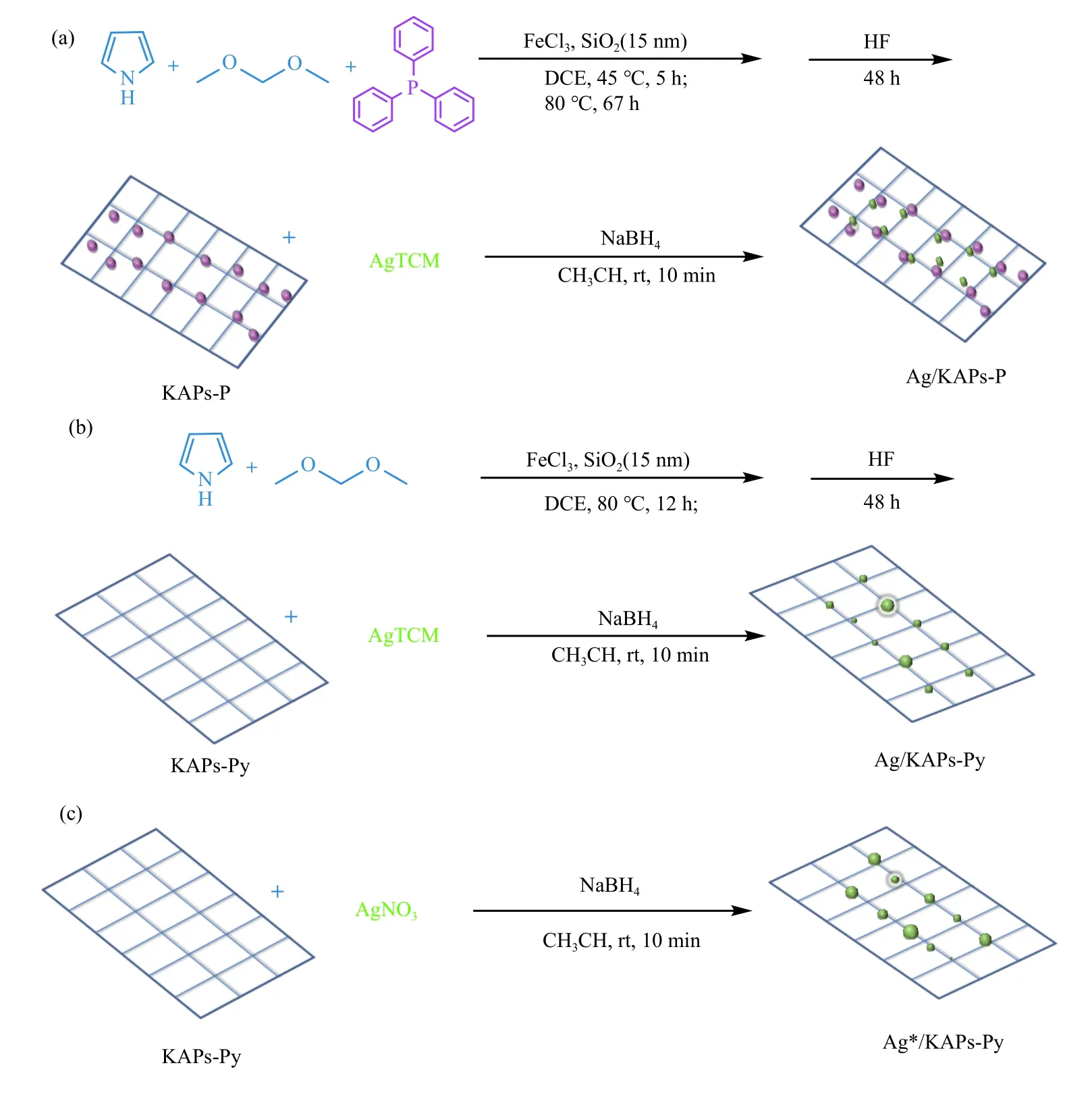
图3 (a)Ag/KAPs-P、(b)Ag/KAPs-Py 和(c)Ag*/KAPs-Py 的合成路径[43]Figure 3 Synthetic route of (a) Ag/KAPs-P,(b) Ag/KAPs-Py,and (c) Ag*/KAPs-Py[43]aMagenta balls represent PPh3 functional groups;green balls represent Ag NPs.Solid line represents boundary of material particles.Dashed lines represent inner network of material particles(with permission from American Chemical Society)
Zhang 等[44]通过鼓泡辅助膜还原方法合成了一种基于三聚氰胺的多孔有机聚合物负载的Ag 催化剂(AgNPs(x)/melamine-based POPs)。当Ag 担载量分别为x=0.9%、2.5%和4.6%时,催化剂中Ag NPs 的平均粒径为5.04、4.33 和6.85 nm。在相同的反应条件下(表1,第8 行),其对应的收率依次为77%、93%、91%。通过N 1sXPS 光谱可检测到随着Ag 的负载量的增加,信号峰向低结合能方向移动,这证实了Ag 和N 之间的相互作用。对比x=0.9%和4.6%的催化剂,虽x=4.6%的催化剂中Ag NPs 具有较大的粒径,但反应活性明显高于x=0.9%催化剂的原因是:x=4.6%的催化剂中Ag 和N 之间的相互作用远强于x=0.9%的催化剂。这也表明,Ag 与N 之间的相互作用对反应活性具有一定的调控作用。
Zhang 等[45]提出氨基诱导的假说去解释载体孔道上不同的氨基官能团对Ag NPs 粒径的调控机制:带有孤对电子的N 可以与Ag+产生强静电相互作用,N 原子周围富电子的区域称为“LAZ(localized active zone)”,LAZ 的体积正比于N 原子在空间中的电子密度,LAZ 的体积越大,对Ag+的捕获能力就越强,形成的Ag NPs 粒径就越大。LAZ 的体积可以由氨基物种的电子供给能力和氨基取代基的空间位阻来调节。氨基物种的电子供给能力越强,位阻效应越小时LAZ 体积越大。具体实验过程如下:对载体OMP[45,55,56]进行氯甲基化、胺化处理引入甲胺(MA)、二甲胺(DMA)、乙二胺(ED)分别形成OMP-MA、OMP-DMA 和OMPED。随后对载体进行负载Ag,各载体上Ag NPs的粒径主要分布为:10.2、7.5、12.7 nm。而后通过制备NOMP 载体实现在载体中引入氨基,随后载Ag 制得Ag@NOMP 催化剂,其中,Ag NPs 的粒径主要集中在4 nm 左右(如图4),符合氨基诱导假说。在最佳反应条件下(表1,第9 行),Ag@NOMP收率最高为96%,其余依次为Ag@OMP-DMA(81%),Ag@OMP-MA (74%)和 Ag@OMP-ED(60%),与此同时反应的TON 也随着Ag NPs 粒径的减小而增大(如图5 所示)。

图4 一锅法合成NOMP 的反应途径,并经过简单“浸渍-还原”法制备Ag@NOMP[45]Figure 4 Illustration of the one-pot synthesis route of amine-incorporated OMP (NOMP),followed by a simple impregnationreduction to give Ag@NOMP[45](with permission from American Chemical Society)

图5 (a)不同催化剂下苯乙炔(EB)与CO2 羧化反应生成苯丙炔酸(PA)的产率(b)Ag 基催化剂中Ag 粒子粒径与TON 之间关系[45]Figure 5 (a) Comparisons of carboxylation of EB with CO2 to PA over various Ag-containing catalysts,(b) Plot for the correlation between the TON of PA produced on Ag catalysts and their particle sizes[45]Reaction conditions: EB (2.0 mmol),catalyst 0.1%,Cs2CO3 (3 mmol),CO2 (1.0 atm),50 ℃,DMSO (15 mL),12 h(with permission from American Chemical Society)
Lan 等[46]使用三聚氰胺和柠檬酸水热法合成MCA 前驱体,随后对前驱体在N2条件下进行热解得到多孔中空的N 掺杂碳管,通过使用乙醇原位还原获得Ag@PHNCT。所制备的Ag@PHNCT具有大量的表面含N 官能团和多孔结构,由于含N 位点对金属纳米颗粒的聚集和浸出的保护,超小的Ag NPs 可以被N 掺杂碳管紧紧地锚定和限制,平均尺寸仅为2.19 nm,这些特性促使了Ag@PHNCT 在炔烃与CO2的羧化反应中表现出优异的催化活性,产率可达98%(表1,第10 行),并且具有优异的稳定性,在五次循环后仍可保持92%的收率,该研究为制备新型催化纳米反应器提供了一种实用有效的策略。
基于上述研究工作可知,Ag NPs 的粒度和分布对CO2的直接羧化具有较高活性。Ag 纳米颗粒由于其高表面能容易在催化过程中聚集[52,57],降低其催化活性和比表面积,导致催化活性和再循环能力降低[51,58],而在载体上引入N、P 等具有孤对电子的元素,利用静电相互作用可以巧妙地锚钉、分散Ag 纳米粒子进而调控粒径大小。
除此之外,Gong 等[36]制备了核壳结构的UiO-66@UiO-67-BPY-Ag 催化剂(如图6)。催化剂以UiO-66 为“核”,采用2,2’-联吡啶-5,5’-二羧酸(H2bpydc)作为壳层UiO-67-BPY 的连接体,连接体中联吡啶基团与Ag 进行配位,将Ag 较好地分散在UiO-67-BPY 壳层中。壳层的厚度调变可以控制传质阻力使反应达到一个比较优异的收率(表1,第11 行),并且壳层起到了稳定活性中心Ag 的目的,在循环五次反应后,循环液中仅检测出0.013%的Ag 析出量。Li 等[47]采用了反相微乳液法制备了核壳结构的xAg@SiO2催化剂,催化剂中SiO2作物理间隔物,以防止Ag 颗粒聚集,并且由于SiO2壳中的大介孔,反应物的扩散屏障可以最小化,使得催化剂具有较高的收率、较大的TOF 值(表1,第12 行)、优异的稳定性和广泛的底物适用性(图7)。

图6 核壳结构的UiO-66@UiO-67-BPY-Ag 合成过程[36]Figure 6 Synthesis of core-shell UiO-66@UiO-67-BPY-Ag[36](with permission from American Chemical Society)
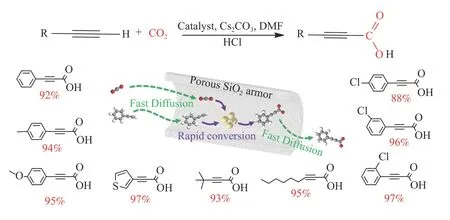
图7 0.2Ag@SiO2 催化炔烃C-H 键与CO2 羧基化反应的底物拓展[46]Figure 7 Substrate scope of 0.2Ag@SiO2[46]Reaction conditions: terminal alkynes (4.0 mmol),catalyst (100 mg),CO2 (1.0 atm),70 ℃,20 h,DMF (5 mL),and Cs2CO3 7.2 mmol(with permission from Elsevier)
此外,结构中引入路易斯碱性物质除了可以起到锚钉Ag NPs 的作用外,还可以与CO2作用,提高催化剂对CO2的吸附能力。Zhang 等[48]报道了一种使用对叔丁基苯胺对氧化石墨烯进行改性从而提高载体对于CO2吸附能力的催化剂设计方案,制备了一系列催化剂:Ag/tert-GO(0.5)、Ag/graphene、Ag/graphite oxide 和Ag/tert-GO-L(表1,第13-16 行),研究表明,酰胺键的存在提高了反应的优异性,其主要归因于以下两方面:对叔丁基苯胺较大的位阻效应可以成功剥离氧化石墨烯形成片层氧化石墨烯,降低了反应底物和活性中心的传质阻力;经过计算得到CO2在tert-GO(接枝酰胺基团的片层氧化石墨烯)上的吸附放热为-6.94 kcal/mol,在GO(氧化石墨烯)上的吸附放热为-4.56 kcal/mol,这也显著地表明酰胺键的生成可以更高效地捕获CO2。理论研究表明,具有三嗪结构的活性N 位点与CO2间存在着某种诱导力,并且CO2的吸附能力与三嗪结构中N 位点的负静电势密切相关[59]。因此,一系列的含有三嗪结构的Ag 基催化剂被广泛应用于该羧化反应。Wu等[49]设计了Ag@NPOP-1 催化剂,催化剂中既含有三嗪结构又含有三唑结构,这一结构导致载体NPOP-1 具有较强的CO2吸附能力,在273 K,0.1 MPa下可吸附CO2达到84.0 mg/g。在不载Ag 时仍然可以促进反应进行,可达55.4%的收率,当载Ag 后收率提升至94.0%(表1,第17-18 行)。除此之 外,CTF-DCE-Ag[50]、Ag0@CTFN[51]、Ag/PCFN-700[52]和Ag@p-CTF-250[53]等含有三嗪结构的催化剂通过三嗪环的N 位点之间的相互作用固定和限制Ag NPs,同时,丰富的氮位点还可以吸附CO2,降低CO2的活化能,在上述催化剂作用下,反应的收率均维持在较高水平(表1,第19-22 行),并且底物适用性广泛。
综上所述,Ag 基催化剂中Ag NPs 与载体的协同催化是保证催化剂优异性能的关键因素。其中,Ag NPs 的小粒径、均匀分散和稳定性是影响反应活性的重要因素,而以上性质均可以通过在载体上引入带有孤对电子的杂原子如N 或P,或者通过物理空间阻隔限域等化学或物理的手段进行控制。同时载体对于CO2的吸附、活化和传质也是重要的因素,载体是一个富电子密度的结构有利于捕获和活化二氧化碳,并且孔道结构对于传质也是重要的,MOF、COF 材料由于其孔结构的可调变性被广泛探究,除此之外一些新颖的催化剂制备方法也可获得具有较低传质阻力的催化剂结构。但是,体系中仍存在一些问题需要解决,如Ag 基催化剂的高负载量和体系的低反应速率。
2.2 Cu 基催化剂
Ag 基催化剂虽然具有较高的反应活性,但是碍于成本较高无法大规模工业推广。Cu 作为非贵金属,来源广泛,且对于端炔与CO2的反应具有催化活性[60,61],因此,Cu 基多相催化剂也被广泛地探究。不同于Ag 基催化剂,在Cu 基催化剂参与下,会发生副反应即炔烃均偶联反应[16,62]。早期CuBr@C[15]、Cunps/Al2O3[63]和 CuCl2@Poly-GLY(1-vim)3(OMs)3[64]等多相催化剂被陆续报道,而这些催化体系均需要有机卤化物的参与去促进反应进行(以上催化剂在相关文献中已被详细介绍,因此,本综述中不再赘述)。本综述将主要介绍近三年来应用于催化端炔与CO2直接羧化的Cu 基催化剂。
Yang 等[61]报道了介孔氮化碳基铜单原子催化剂(Cu-CN-x),Cu 掺杂在氮化碳骨架中代替了其中一个碳原子与三个N 原子相连,其中,Cu 的价态为+1 价。在一系列不同Cu 担载量的催化剂中,担载量为8%的催化剂具有最好的反应活性,收率可以达到97%,TOF 可达到9.7 h-1(表2,第1行),并且催化剂具有较好的稳定性,在五次循环过程中收率几乎没有变化,并且在反应溶液中也没有检测到浸出的Cu 物种。随后基于DFT 分别对CN 和优化后的Cu-CN-8 计算CO2插入脱质子的苯乙炔所需的能量:在CN 中,脱质子的苯乙炔与N 相连,在插入CO2后,羧基的氧原子更倾向与N 邻近的O 原子相连。而这一过程活化能为+2.31 eV,需吸热1.39 eV。但Cu-CN-x催化剂上二氧化碳插入Cu-C(sp)的活化能为+0.85 eV,最终放热0.27 eV,见图8。可见Cu 物种的引入促进了中间体的形成和稳定。
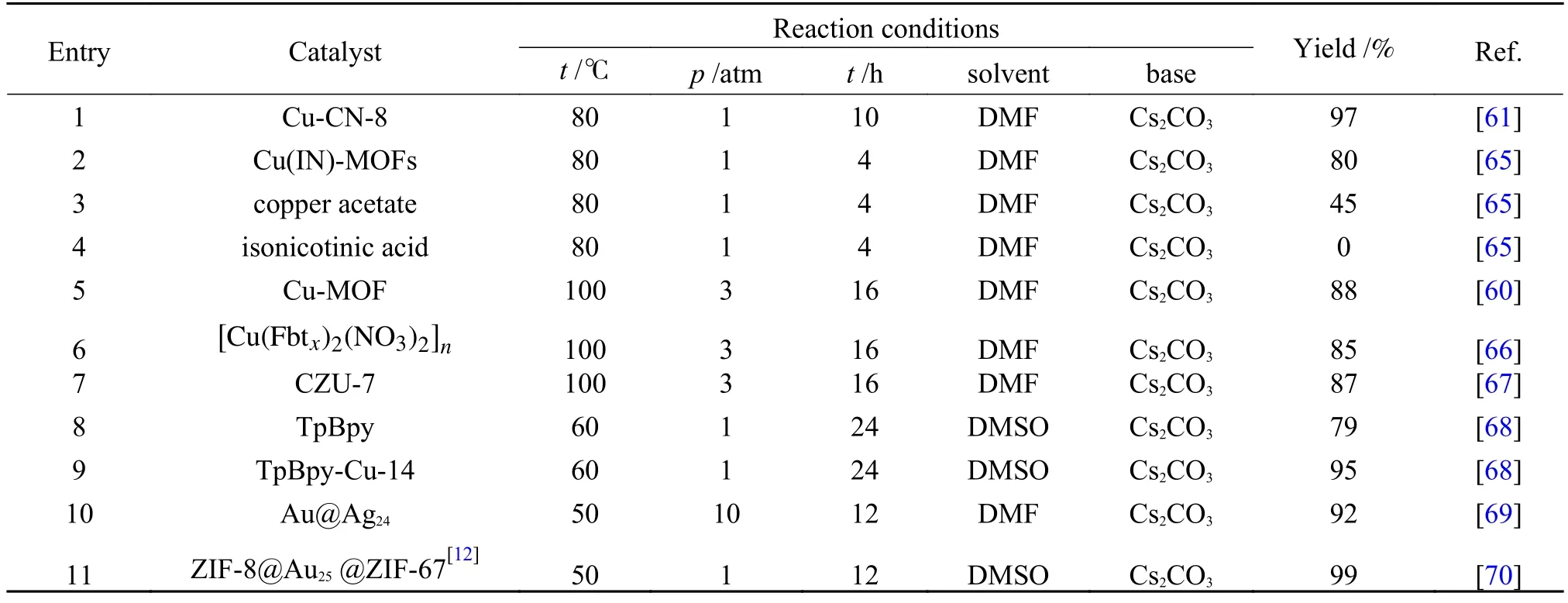
表2 端炔与CO2 直接羧化反应的Cu 基与其他多相催化体系Table 2 Heterogeneous catalytic system for direct carboxylation of terminal alkynes with carbon dioxide

图8 CO2 插入Cu-CN-8.0 和CN 表面上脱质子化的苯乙炔中间体的反应路径(插图:Cu-CN-8.0 的优化结构)[61]Figure 8 Reaction profile for CO2 inserting into the deprotonated phenylacetylene intermediate on Cu-CN-8.0 and CN surfaces (inset: the optimized structure of Cu-CN-8.0)[61](with permission from American Chemical Society)
同时,Cu 位点所处的化学环境对活性有着重要的影响。Cu(IN)-MOFs[65]是由醋酸铜和异烟酸通过水热法进行合成的二维平板形貌的配位聚合物。异烟酸是一种既含有吡啶基又含有羧基的双功能配体,Cu(II)同时与吡啶基的N 和羧基的O 配位去平衡电荷。有趣的是,该催化剂在第一次反应时收率为80%(表2,第2 行),而第二次循环时收率可以达到83%。通过XRD 和XPS 表征发现了使用过的催化剂结构上的变化。这种变化主要表现在两个方面:使用过的催化剂中Cu(I)的位点增多;结构中的N 位点更多的暴露。由此推断出N-Cu 键为弱配位,在催化过程中会发生NCu 键的断裂,而Cu(I)的催化活性要优于Cu(II),具体催化过程如图9。同时MOF 结构中吡啶基N 位点可以与Cu 位点结合用做助催化剂,增强CO2的吸附能力。为了证明MOF 前体不产生催化活性,研究人员分别使用醋酸铜和异烟酸做催化剂去进行反应后发现产率都非常低(表2,第3-4 行),表明了Cu(IN)-MOF[60]结构是显著影响催化活性的。除此之外,采用微波辅助合成的配位聚合物如Cu-MOF、[Cu(Fbtx)2(NO3)2]n、[Cu(Fbtx)2(BF4)2]n、[Cu(Fbtx)2SO4]n[66]和 CZU-7([Cu(Fbtx)2(Br)2]n)[67],这些配位聚合物也被广泛应用于端炔与CO2的羧化反应,并维持一个较优的产率(表2,第5-7 行),同时这些配位聚合物中任何一个结构单体都不具备高效的催化活性,只有当它们合成配位聚合物时才具有高活性。值得一提的是这些聚合物具有很好的鲁棒性,在多次循环后,产率没有明显变化,并且耐酸碱能力较好。

图9 Cu(IN)-MOF 催化端炔与CO2 羧化反应的机理[65]Figure 9 Proposed reaction mechanism based on the Cu(IN)-MOF catalyzed carboxylation of terminal alkynes with CO2[65](with permission from Elsevier)
又如Bu 等[68]制备了一种Cu(I)修饰的COF 催化剂,该COF 材料是由Tp 与Bpyda 制备而成(图10)。Cu(I)与TpBpy中的N、O 形成的螯合位点进行配位从而固定在载体上。TpBpy 结构中的N、O 位点可以与端炔的C-H 键形成氢键,从而起到活化C-H 键的作用,而且结构中的富N 区域由于其高电子密度可以很好地吸附CO2,这些优异的性能体现在当只有TpBpy 加入时,催化收率可达79%(表2,第8 行)。而Cu 的引入也显著提高了反应活性,在Cu 含量为最佳负载时,反应的收率提高到95%,TOF 值为45 h-1(表2,第9 行),催化性能的提高可归因于催化剂中的Cu(I)可以与C≡C 发生ɳ2配位活化炔烃的C-H 键,在碱的作用下,更容易脱去质子,与此同时,形成的炔铜结构也更有利于CO2亲电进攻形成炔酸铜。但该催化剂在循环三次之后,收率会有明显的下降。下降的原因主要为:在反应过程中Cu(I)被氧化成Cu(II)。当循环了三次的催化剂使用KI 处理后,反应的活性得到恢复(图11)。为了提高催化剂的可循环性,研究人员提出可以通过将较软的路易斯碱位点引入载体中,达到稳定Cu(I)的目的。

图10 TpBpy 结构示意图[68]Figure 10 Schematic representation of TpBpy[68](with permission from Elsevier)

图11 (a)TpBpy-Cu-14 催化剂的循环测试性能图;(b)未使用的、使用过的和再生的催化剂的Cu 2p XPS 谱图[68]Figure 11 (a) Recycling tests of the TpBpy-Cu-14,reaction condition: CO2(1 atm),1-Ethynylbenzene (1 mmol),catalyst(10 mg),Cs2CO3 (1.5 mmol),6 h,60 ℃,(b) Cu 2p XPS spectra of the fresh,used and regenerated catalysts[68](with permission from Elsevier)
综上所述,Cu 基催化剂在端炔与CO2的羧化反应中具有较好的活性,其中,Cu(I)相较于其他氧化态具有更好的催化活性。Cu 在反应过程中通过与炔烃的C≡C 进行配位,从而达到活化炔烃的作用,同时炔铜中间体的生成也更利于CO2亲电进攻生成炔酸铜。其中,Cu 所处的化学环境直接影响到催化剂的稳定性和催化剂的活性,当软碱与Cu 配位时Cu(I)的氧化态可以被更好地保护,Cu 配位能力的强弱影响Cu 的浸出能力,催化剂的电子密度也会直接关系到CO2的吸附能力。相较于Ag 基催化剂可以发现,Cu 基催化剂的催化活性相对较低,并且需要较高的Cu 担载量和较高的反应温度,因此,更加高效的Cu 基催化剂仍有待开发。
2.3 Au 基催化剂
部分Au 基催化剂也被应用于端炔与CO2的直接羧化反应,如Liu 等[69]报道了在Ag25团簇中掺杂Au、Pd、Pt 形成Au@Ag24、Pd@Ag24和Pt@Ag24。其中,反应活性顺序如下:Au@Ag24>Pd@Ag24≈Pt@Ag24>Ag@Ag24。这种活性差异是由于外来杂原子的参入影响团簇的电子结构而产生的[69]。再如类似三明治结构的复合催化材料ZIF-8@Au25@ZIF-67[tkn] (tkn=thickness of shell)(图12)[70]。具体制备如图13,通过物理浸渍将Au25(LCys)18的溶液缓慢倒入ZIF-8 溶液中,以形成纳米材料Au25/ZIF-8,随后将特定浓度的Co(NO3)2缓慢添加到Au25/ZIF-8溶液中,通过Co2+和羧酸基团之间的配位,在Au25/ZIF-8 表面形成Co2+涂层。将2-MeIm 溶液缓慢添加到上述体系中即制得夹层复合材料ZIF-8@Au25@ZIF-67,其中,外壳层ZIF-67 的厚度(即[tkn])可以通过调变Co2+和2-MeIm 的浓度来控制。

图12 ZIF-8@Au25@ZIF-67[tkn] 和 ZIF-8@Au25@ZIF-8[tkn]的合成路径[tkn=壳层厚度][70]Figure 12 Synthetic Route for the Sandwich Structures of ZIF-8@Au25@ZIF-67[tkn] and ZIF-8@Au25@ZIF-8[tkn] [tkn=Thickness of Shell][70](with permission from American Chemical Society)

图13 (a)不同催化剂催化苯乙炔羧化的反应活性;(b)三种不同催化剂的柱状图;(c)具有不同壳层厚度的ZIF-8@Au25@ZIF-67 催化性能折线图;(d)不同催化剂的TPD-CO2[70]Figure 13 (a) Catalytic activity of various catalysts for the carboxylation of phenylacetylene.Reaction conditions: catalyst (1.12 ×10-4 mmol of Au25),alkyne (0.5 mmol),Cs2CO3 (0.24 mmol),CO2 (1.0 bar),50 ℃,12 h,(b) Column diagram of three different catalysts,(c) Broken line diagram of catalytic performance of ZIF-8@Au25@ZIF-67 with various shell thicknesses,(d) TPD-CO2 by various catalysts[70](with permission from American Chemical Society)
实验证实,通过控制壳层厚度,催化剂的催化性能得到了精确优化(图14),其最佳的壳层厚度为12 nm,此时所对应的收率为99%(表2,第11行),并且催化剂具有良好的稳定性,在三次循环后催化剂的结构几乎未发生变化。随后研究人员分别探究了ZIF-8、ZIF-67、ZIF-8@Au25@ZIF-67[12]和ZIF-8@Au25@ZIF-8[12]对于CO2的吸附能力,发现ZIF-67 与ZIF-8@Au25@ZIF-67[12]吸附能力相近,ZIF-8 和ZIF-8@Au25@ZIF-8[12]吸附能力相近,由此得出壳层物种直接影响CO2的吸附,其中,ZIF-67 具有非常优异的CO2吸附能力(图14)。因此,ZIF-8@Au25@ZIF-67[12]催化性能的优越离不开结构中的壳层,壳层在这里面起到了控制传质阻力的效果、稳定Au 活性物种的作用和优异的CO2的吸附能力。
3 反应机理
多相催化炔烃C-H 键与CO2羧基化反应体系中普遍认同的反应机理如下:在碱和金属存在下,末端炔烃脱质子与金属(M)配位形成M-C 键,产生金属炔化物中间体;随后CO2插入M-C 形成R≡C-COO-M;最后在碱的作用下,生成炔酸盐从催化剂上脱附,催化剂再生。有计算表明,金属炔化物是该反应的重要中间体,速率决定步骤是CO2亲电进攻金属炔化物生成R≡C-COO-M[37]。其中,Ag 基催化剂整体的反应活性要比Cu 基要高,根据DFT 计算均相(NHC)2-Ag 和(NHC)2-Cu体系表明,这种反应优势可能归因于Ag-C 键比Cu-C 键要长,更早形成过渡态且该过程的自由能垒较低[71]。
反应体系中的碱具有打破热力学平衡限制的重要作用。碱的存在有利于端炔去质子生成炔化物中间体[69,71],同时碱性物质的碱性强度会影响反应的产率,且碱性物质的组成也会在反应过程的中间体中出现。关于碱的种类有:Cs2CO3、K2CO3、Na2CO3,KOtBu、NaOtBu,NaOH、KOH 等无机碱和1,8-二氮杂二环[5.4.0]十一烯-7-烯(DBU)、DMF和TMEDA 等有机碱。其中,Cs2CO3是目前活性最好的碱物质,其pKa 在10.33 左右、端炔的pKa大致在25,的pKa 在10 左右,因此,端炔是不可能直接被Cs2CO3脱质子的。依据这些考虑,提出金属活性中心先与炔烃进行配位,增强端炔氢的酸性,随后在Cs2CO3的帮助下脱除质子[72]。DMF 作为一种弱的路易斯碱[65],常用作反应体系的溶剂,一方面促进CO2和Cs2CO3的溶解;另一方面可以脱除端炔氢促进M-C 键的生成。这也解释了在无Cs2CO3存在下,反应体系以DMF 为溶剂时会有少量的底物转化,但在以DMSO 作为溶剂时,反应几乎不发生。
体系的溶剂效应也被探究,据报道DMF、DMSO 或碳酸亚丙酯(PC)是有利的溶剂,不同催化剂中最优溶剂的选择不同。有机碳酸盐(DMC、DEC 和PC)可以稳定中间体如乙炔化铜,并加强反应物种间相互作用[64,73]。更强的极性溶剂(DMSO)可以促进Cs2CO3在液相中的溶解[43],另外,Wu 等[54]依据DFT 分别计算了在DMSO 和DMF 中CO2亲电进攻金属炔化物中间体这一过程中的能量变化,在DMSO 溶液中,形成过渡态所需能垒∆EDMSO=15.1 kcal/mol,而在DMF 中该过渡态能垒为∆EDMF=16.1 kcal/mol。因此,从热力学角度考虑,DMSO 这一极性较强的溶剂对于反应更有利,由此可知,极性溶剂不仅对Cs2CO3有较好的溶解度还可以稳定羧化反应的极性过渡态络合物[68](表1,第24-25 行)。
丙炔酸金属中间体对于温度较为敏感,在温度升高时将会发生脱羧反应[29,68]。如Dang 等[50]的研究中发现反应温度在50 ℃为宜,过高的温度会导致脱羧,而较低的温度下相同时间内产物的产率较低。Bu 等[68]探究了反应的动力学。动力学研究显示该反应的动力学为一级反应动力学。在50 和60 ℃时对应的速率常数分别为0.00097 s-1和0.002 s-1。可见在一定的温度范围内,温度对于反应速率的影响较大。因此,在端炔与CO2的羧化反应中温度的选择至关重要,存在一个最佳反应温度,该最佳反应温度应在满足热力学限制的条件下尽可能去提高动力学上的反应速率。另外,部分研究者在反应过程中引入卤代烷[15,63,74],在丙炔酸产物生成的同时实现其原位酯化(carboxylation of terminal alkyne and esterification,CTA-E),再通过二次水解获得酸类产物[75](表3),通过酯化移除羧酸盐产物,使反应平衡正向移动。
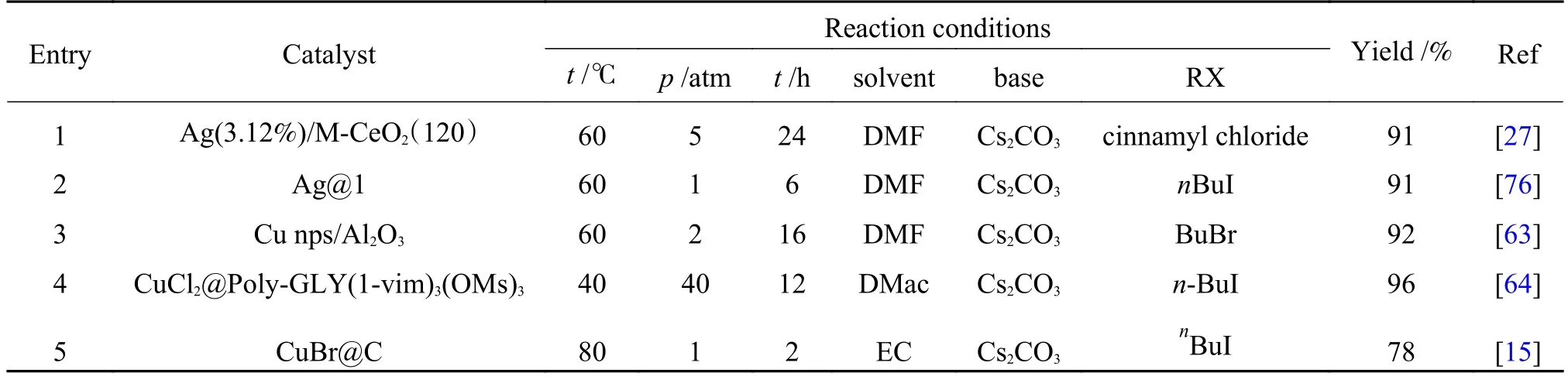
表3 端炔与CO2 的在卤代烷(RX)作用下的羧化反应Table 3 Carboxylation of terminal alkyne with carbon dioxide in the presence of haloalkanes
根据文献报道,该反应体系对水是高敏感性的,对此有研究从反应机理的角度给出了解释,由于水的酸性强于端炔氢,因此,在水存在下会存在端炔与水竞争脱氢,Cs2CO3会优先与H2O 进行脱质子生成CsHCO3,而非脱去端炔的质子。并且水对于反应收率的影响与Cs2CO3与水的比例密切相关,当反应体系中只有极其微量的水存在,Cs2CO3可以将其“吸收”后仍有满足反应所需的碱量时,反应可以正常进行,但是如果体系中含有较多的水,从而消耗大量的碱,剩余的碱量不足以促进反应进行时,反应的产率将明显降低(表4)[77]。综上所述,基于反应机理,总结了碱的作用机制、溶剂效应、温度和水对于反应体系的影响。
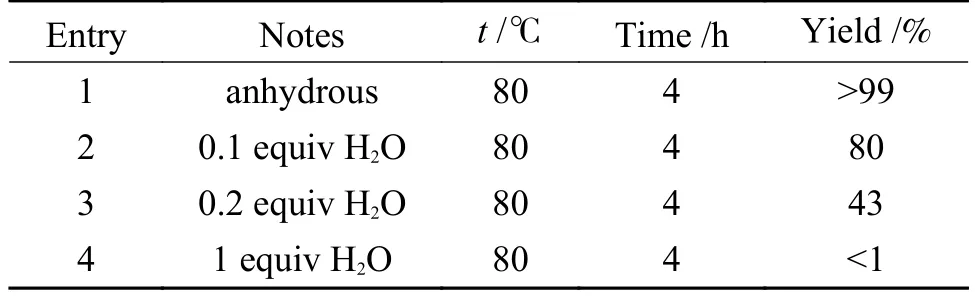
表4 水在无金属催化时对于反应的影响Table 4 Effect of water on the catalyzed and catalyst-free reaction
4 总结与展望
本工作从反应体系的活化、催化剂和反应机理三个角度进行综述目前端炔与CO2的羧化反应进展。端炔具有两种活化方式:端炔的C≡C 与金属(Ag、Cu 和Au)配位后,在碱的作用下进行脱质子;催化剂中N、O 元素与端炔氢形成氢键极化C-H 键,随后在碱的作用下脱质子。二氧化碳的活化则是通过设计富电子结构的催化剂。在此基础上总结了目前催化剂设计的主要观点:控制金属活性中心的尺寸和分散程度;通过控制载体上的电子密度和引入官能团来增强载体对于底物的吸附和活化能力;设计载体结构降低传质阻力。活性中心与载体协同催化提高反应的催化效率。并总结了目前公认的一种反应机理:在碱和金属存在下,末端炔烃脱质子与金属(M)配位形成M-C 键,产生金属炔化物中间体;随后CO2插入M-C 形成R≡C-COO-M;最后在碱的作用下,生成炔酸盐从催化剂上脱附,催化剂再生。其中,金属炔化物是该反应的重要中间体,速率决定步骤是二氧化碳亲电进攻金属炔化物生成R≡C-COO-M。
尽管通过这些催化剂制备方法和反应机理的探究,贵金属和非贵金属催化剂都取得了一些优异的结果,对于反应机理也有了一定的认知,但仍存在一些问题,需要在未来解决。
首先,CO2在热力学和动力学上的惰性导致端炔与CO2直接羧化反应即使在较苛刻的条件下反应速率仍较慢。虽然目前的多相催化体系使得反应在相对温和的条件下进行,但是其反应速率较低的瓶颈仍未能突破。为此更加高效的催化体系有待被设计开发。目前,主要的催化活性金属为Ag、Cu 和Au,其中,Au 基催化剂研究相对较少,Ag 基催化剂具有相对较好的催化活性,而Cu 相对次之。但是由于Ag 和Au 相对昂贵,若想将该反应发展到工业应用的水平,还需要提高Cu 基催化剂的活性和拓宽适用于该反应的催化活性金属。根据目前的反应机理探究表明,反应的速率决定步骤为CO2插入M-C 键,同时Cu、Ag、Au 位于化学周期表中的同一主族,在扩宽活性金属时可以适当考虑与这三种元素性质相近的金属元素。
其次,反应机理仍需深入探究。反应需要碱性助剂的引入帮助端炔脱质子,目前,最高效的碱为Cs2CO3。但是尚未有研究明确表明Cs2CO3为何具有较高的活性,仍需探究一下这种高效性是由于其碱性上的相对优势,还是由于其在既有碱性优势的同时Cs+在后续反应中也会作为某种助剂对于反应具有催化活性,这需要从反应机理的角度进行探究。同时对于Cs2CO3的探究也将为反应碱性物质的选择提供基础。
最后,体系对于水是高度敏感的,依据目前的知识,这种敏感性一方面是由于碱性物质会与水发生反应;另一方面可能是由于反应中间体金属炔化物对水的高敏感性导致的,因此,设计不含活性金属的催化剂对于摆脱体系对于水和氧气的高敏感性具有重要的研究意义。
猜你喜欢
上海师范大学学报·自然科学版(2023年1期)2023-06-30
能源化工(2021年2期)2021-12-30
中国有色金属学报(2018年6期)2018-07-09
石油炼制与化工(2017年1期)2017-04-06
赣南师范大学学报(2016年3期)2016-07-18
广东石油化工学院学报(2016年6期)2016-05-17
合成化学(2015年5期)2015-03-26
化工技术与开发(2015年6期)2015-01-29
石油炼制与化工(2014年4期)2014-04-06
石油化工应用(2014年2期)2014-03-11
