
小麦DNA去甲基化酶基因全基因组鉴定及其在籽粒发育中的表达分析
2023-10-23 08:18蒋正宁刘大同李东升程晓明高德荣
麦类作物学报 2023年11期
蒋正宁,刘大同,张 晓,江 伟,王 玲,李东升,程晓明, 高德荣
(江苏里下河地区农业科学研究所/农业部长江中下游小麦生物与遗传改良重点实验室,江苏扬州 225007)
DNA甲基化是生物体内一种重要的表观遗传修饰手段,在高等真核生物中,DNA 甲基化发生于胞嘧啶第五位碳原子上形成 5-甲基胞嘧啶(5-methylcytosine,5-meC)[1],DNA甲基化状态是由甲基化建立、维持和主动去甲基化动态调节的结果[2],而DNA 去甲基化则是 DNA 甲基化的相反过程,即 5-甲基胞嘧啶被未修饰的胞嘧啶替代的过程[3]。在植物中,DNA 去甲基化包括 DNA 主动去甲基化和被动去甲基化2 种类型:被动的去甲基化,是由于 DNA 复制过程中甲基转移酶活性的丧失或甲基供体的缺乏导致甲基化无法维持,从而使整个基因组 DNA 甲基化水平降低;DNA 主动去甲基化,则不依赖于DNA复制,即通过5-甲基胞嘧啶 DNA 糖基化酶作用,直接去除 DNA 中甲基化的胞嘧啶[4]。DNA去甲基化不仅对于全基因组表观遗传重编程至关重要,在植物发育过程中还介导转录因子或基因座特异性基因激活[4]。目前,普遍认为DNA去甲基化的机制有 5 种:依赖DNA 去甲基化酶的碱基切除修复机制、碱基切除修复、甲基胞苷脱氨基耦合G/T 的错配切除修复、水解作用去甲基化以及氧化去甲基化[5]。在所有的机制中DNA的去甲基化是通过DNA去甲基化酶(dMTase)实现的,DNA去甲基化酶含有最保守的DNA 糖苷酶(HhH-GPD)结构域,具有糖基化酶和脱嘌呤裂合酶活性的双功能。
目前,在植物中已鉴定6种dMTase,包括转葡萄糖基酶 DME ( demeter) 、 沉默抑制因子 ROS1 (repressor of silencing 1), 以及 类转葡萄糖基酶蛋白 DML2(demeter-like protein 2) 、DML3、DML4和类转葡萄糖基酶DML5[6]。研究表明ROS1和DML在所有的营养组织中表达,包括根和茎的组织,而DME主要在种子发育中表达[7]。在拟南芥中,DME是孢子体发育所必需,并在孢子体生命周期中维持干细胞活动起着至关重要的作用[8]。此外,ROS1和DME也参与调控水稻[9]和大麦的种子发育[10]。最近,3个水稻dMTase基因(DNG702、DNG701和DNG704)被证实可以对配子体和合子细胞的DNA去甲基化,是合子基因表达和发育所必需[11];番茄DNA去甲基化酶基因SlDML2与其果实成熟过程中DNA甲基化水平普遍降低密切相关[12]。上述研究表明,dMTase对植物合子发育和果实成熟起到重要的调控作用。
此外,植物的生物或非生物应激反应也需要DNA主动去甲基化的调控。例如,拟南芥可以通过ROS1对RMG1和RLP43基因启动子的去甲基化激活抗病反应[13],而其DNA 去甲基酶三重突变体rdd(ros1/dml2/dml3)对镰刀形孢菌表现高度敏感[14];在水稻中,盐敏感品种IR29在盐胁迫下诱导dMTase基因(DNG701和DNG710)表达[15];而在烟草中过表达AtROS1,则增加了类黄酮生物合成途径相关基因和抗氧化途径相关基因的去甲基化水平,激活了这类抗逆基因的表达,从而导致盐胁迫耐受性提高[16];此外,DNA去甲基化也在植物热和干旱胁迫反应的调控中发挥作用[17-18]。可以看出,DNA去甲基化酶基因在调节植物的各类生物学过程中发挥重要作用。
目前,DNA去甲基化酶(dMTase)基因在很多物种中被广泛鉴定和分析,这些物种中dMTase 的数量、保守性、多态性和功能都存在很大差异。作为重要粮食作物的小麦dMTase基因家族成员尚未被鉴定。本研究基于小麦全基因组测序数据,利用生物信息学方法对小麦dMTase基因家族进行了全基因组鉴定,分析小麦dMTase基因核苷酸及编码蛋白的特性及其组织特异性表达模式,并通过转录组测序及qRT-PCR方法分析小麦dMTase基因在籽粒发育中的表达特征,为进一步解析小麦dMTase基因功能研究奠定基础。
1 材料与方法
1.1 小麦dMTase 基因家族成员的筛选
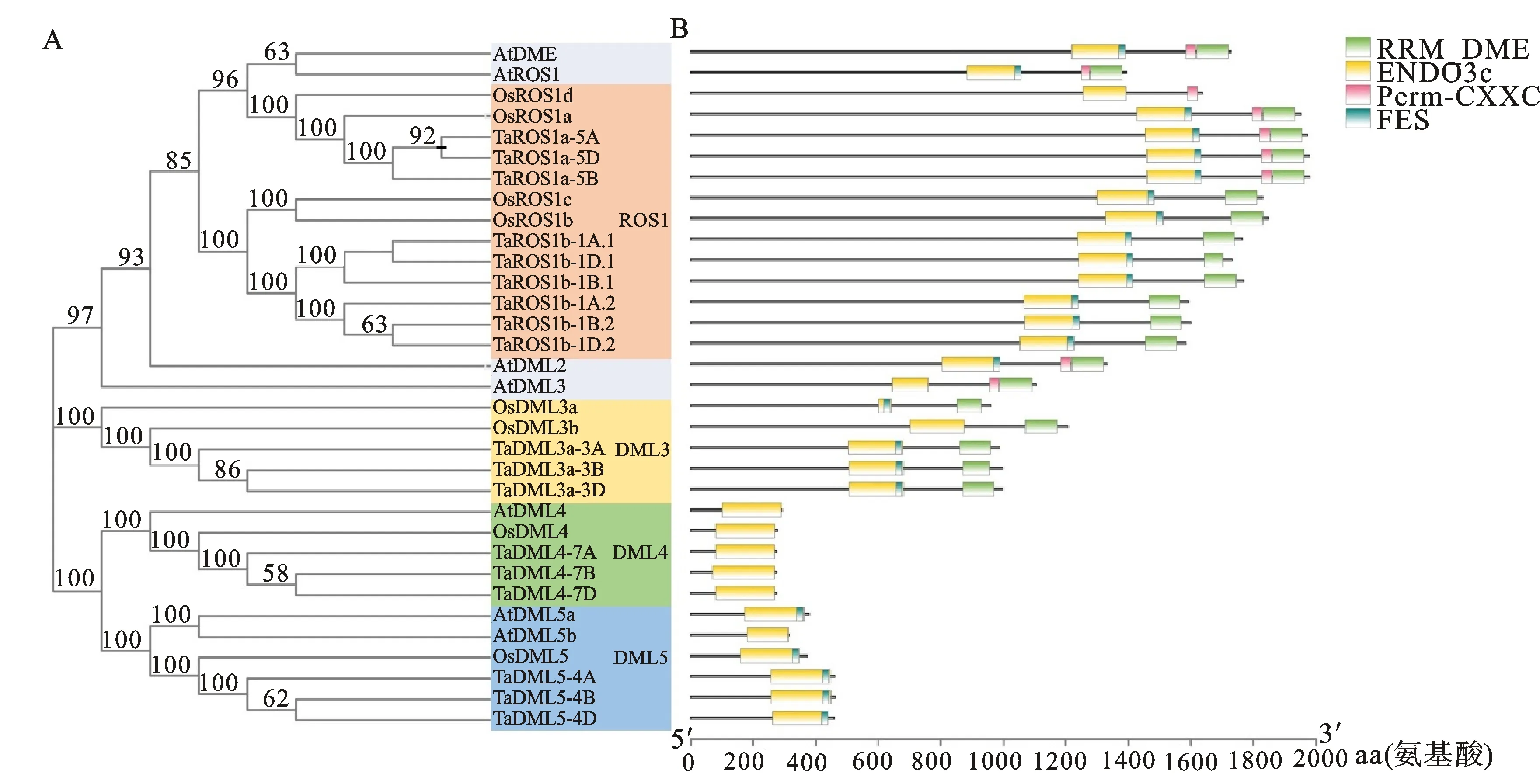
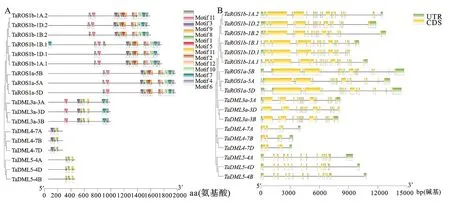
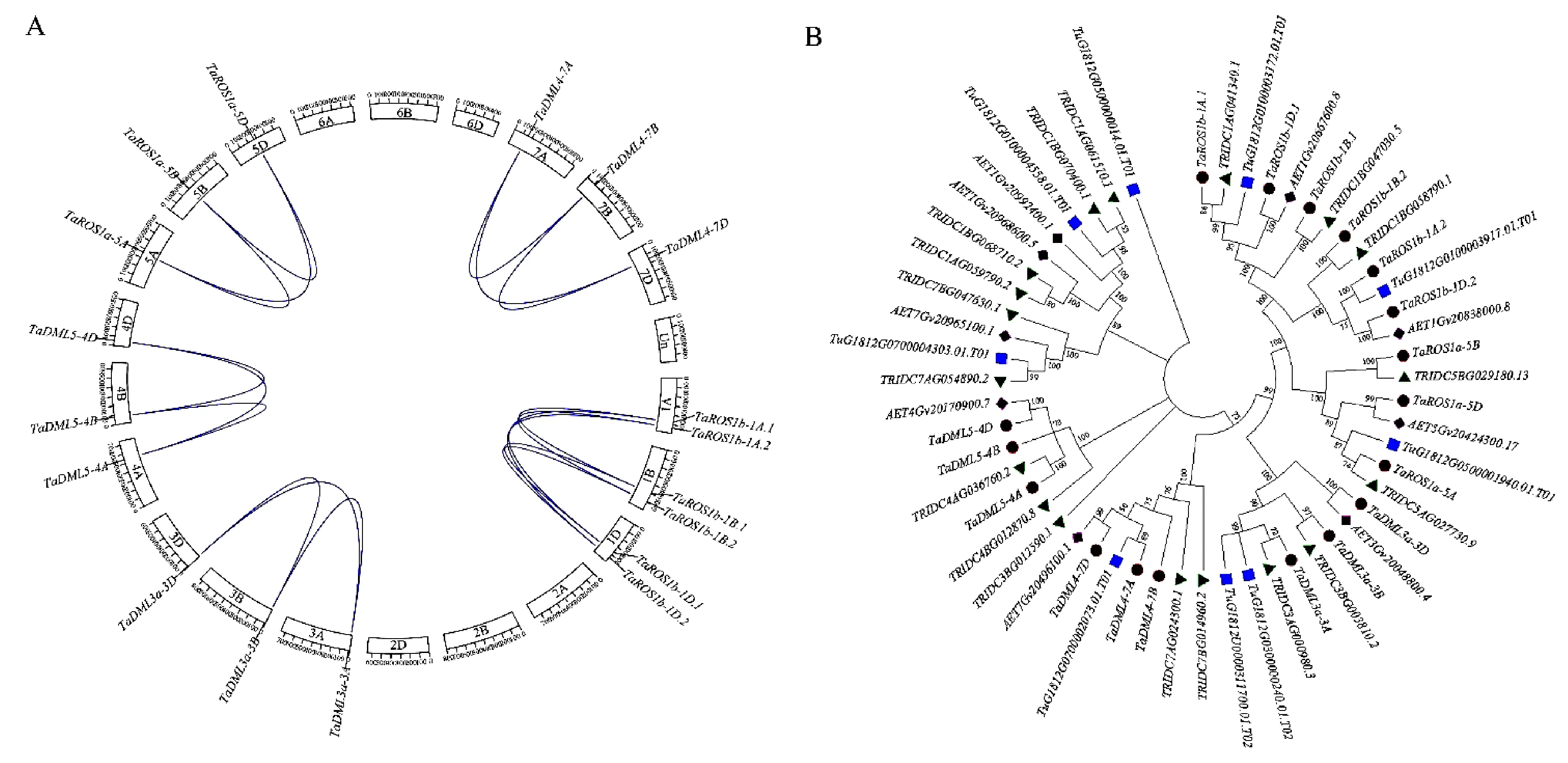
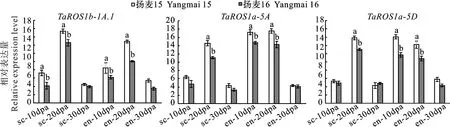
从Ensembl Plants数据库 (http://plants.ensembl.org/index thml )下载小麦蛋白序列数据、基因组序列和注释文件,并建立本地数据库。以已知的拟南芥[14]和水稻[19]dMTase基因家族成员蛋白序列作为query 序列,与小麦的蛋白序列进行本地BlastP比对,筛选阈值设为E 利用ExPASy 网站(http://expasy.org/)对小麦dMTase氨基酸序列进行等电点、分子量预测等理化性质分析;利用Plant-mPLoc(http://www.csbio.sjtu.edu.cn),对小麦dMTase进行亚细胞定位分析;根据 CDD(https://www.ncbi.nlm.nih.gov/cdd/)所提供的家族成员所含结构域起始位点整理结构域氨基酸序列。利用MEME (http://meme-suite.org/tools/meme) 在线预测dMTase家族蛋白的保守motif,最大发现数设为12个,其他参数设为默认值。使用TBtools[20]软件对结构域及保守motif位点进行可视化显示。 从Ensembl Plants数据库中下载拟南芥和水稻dMTase的氨基酸序列,使用Clustal W 软件进行dMTase同源蛋白的氨基酸序列比对。利用MEGA 11(https://www.megasoftware.net/)软件构建系统进化树,选择邻接法( Neighbor-joining ),bootstrap 方法重复采样1 000次,其余参数默认,参考拟南芥和水稻 dMTase基因家族的亚族分类结果对小麦dMTase 基因家族进行亚族分类,并依据小麦dMTase染色体位置进行基因命名。利用ITOL(https://itol.embl.de)在线软件对进化树进行美化处理。 从Ensembl Plants数据库中下载的小麦基因信息gff3文件中提取dMTase基因染色体位置和基因结构信息,利用GSDS 2.0( http://gsds.cbi.pku.edu.cn )分析小麦 dMTase基因的外显子和内含子分布模式。从相同数据库中下载小麦基因组(DNA)数据,利用TBtools 软件截取小麦dMTas基因起始密码子上游2 000 bp的DNA序列,利用Plant CARE(http://bioinformatics.psb.ugent.be/webtools/plantcare/html)工具分析启动子顺式作用元件种类、数目及功能,并使用 TBtools 软件进行可视化。 根据小麦基因组注释gff3文件获取dMTase基因家族成员的染色体位置信息,利用TBtools 软件的MCScanX工具计算并获取小麦dMTase共线性及基因的串联重复信息,并进行对小麦基因的染色体位置信息、共线性关系、串联重复信息进行可视化。利用TBtools软件计算直系同源基因间的同义替换率(Ks) 和非同义替换率(Ka)。通过Ka/Ks 值来评估小麦dMTase 基因在进化过程中受到的选择压力影响,Ka/Ks>1、<1或=1分别表示复制基因对受到正向选择、纯化选择和中性进化的影响。 从Wheat Exp数据库(http://www.wheat-expression.com) 检索小麦RNA-Seq数据,用TPM(transcripts per million reads)值LogScale标准化后评估小麦基因的转录本丰度值。用TBtools软件绘制小麦TadMTase基因表达热图。 表1 荧光定量分析引物Table 1 Primers used forreal-time PCR 以7个拟南芥和8个水稻dMTase蛋白为参考序列,结合BlastP和HMM 模型Pfam(PF00730)HMMER3.0两种方法搜索小麦dMTase蛋白序列,之后进行序列比对,去除冗余序列后合并,利用Pfam和SMART结构域搜索检验候选基因是否含有ENCO3c结构域(ENCO3c包含 HhH-GPD),最终在小麦基因组中共鉴定出18个候选基因,并根据系统进化关系和其染色体位置进行命名。在ExPASy数据库中对小麦所有dMTase蛋白序列进行理化性质预测分析,结果(表2)表明,小麦dMTase基因家族蛋白氨基酸长度为276 ~1 982个aa,分子量为30.7~ 218.4 kDa。蛋白等电点为6.0 ~ 9.7,均值为7.48,预测大部分为中性蛋白;小麦 dMTase蛋白不稳定系数均大于 40,为不稳定蛋白;蛋白亲水性(GRAVY)分析显示小麦所有 dMTase 蛋白都是亲水性蛋白。亚细胞定位预测表明,小麦dMTase基因编码蛋白均定位于细胞核中(表2)。 将检索到的 18个小麦dMTase基因的蛋白序列,与已知的7个拟南芥dMTase基因[AtDML5b(AT1G05900.1)、AtDML5a(AT2G31450.1)、AtROS1(AT2G36490.1)、AtDML2(AT3G10010.1)、AtDML4(AT3G47830.1)、AtDML3(AT4G34060.3)、AtDME(AT5G04560.1)]和8 个水稻 dMTase 基因[OsROS1a(Os01g11900.1)、OsROS1d(Os02g29230.1)、OsDML3a(Os02g29380.1)、OsDML3b(Os04g28860.1)、OsROS1b(Os05g37350.1)、OsROS1c(Os05g37410.1)、OsDML4(Os06g13070.1)、OsDML5(Os11g16580.1)]共计33 个dMTase基因的蛋白序列进行比对,并构建系统进化树。基于水稻OsdMTase分类,将小麦18个dMTase归属于ROS1、DML3、DML4和DML5等 4个亚家族。其中ROS1最多,包含9个dMTase基因,其他亚家族各有3个(图1A)。利用 CDD 对TadMTase进行保守结构域分析,结果(图1B)表明,所有的TadMTase均含有ENCO3c保守结构域,ENCO3c结构域行使糖苷酶功能是dMTase 特异性结构域。TadMTase不同亚族所包含的保守结构域种类不同,ROS1和DML3亚族包含3 个共同保守结构域:ENCO3c(包含 HhH-GPD domain)、FES以及RRM_DME结构域。其中小麦TaROS1a-5A/B与拟南芥AtROS1、水稻OsROS1a的ROS1亚家族还包含了一个介导 DNA 结合活性的Perm-CXXC 结构域。而DML5包含ENCO3c和FES 2个结构域和DML4仅包含1个 ENCO3c结构域。 图1 TadMTase 进化树(A)和保守结构域(B)分析 小麦18个DNA去甲基化酶的motif分析如图2A所示,小麦DNA去甲基化酶基因包含了12个motif结构,但不同亚族的所拥有的Motif不同,与其保守结构域一致。如ROS1亚家族的motif组成相同,包含motif 1、2、3、5、6、7、9 、10和11共9个保守motif,组成ENDO3c、FES、Perm-CXXC、RRM_DME 结构域,其中motif1、6、7和9共同构成一个保守的ENDO3c糖苷酶结构域。而DML3亚家族包含7个motif,分别是motif 1、2、3、5、7、9和11,它们构成了ENDO3c、FES和RRM_DME结构域;DML4亚家族仅包含2个motif结构,分别是motif 1和7,它们构成了ENDO3c结构域;DML5亚家族包含4个motif结构,即motif1、2和9构成了ENDO3c和FES结构域。 图2 TadMTase 基因家族保守基序(A)和基因结构(B) 为进一步阐明小麦TadMTase基因的结构特征,利用 GSDS 2.0研究基因的外显子-内含子结构,分析显示TadMTase亚组间基因结构差异较大,其中ROS1包含18~22个外显子(图 2B),DML3包含17个外显子,DML5包含13个外显子,而DML4只包含4个外显子。这与已报道的植物dMTase基因结构相似,进一步说明虽不同物种在进化过程中发生了分化事件,但其同源基因在分化后仍具有相似结构,进一步说明dMTase家族具有其保守性。 利用 Plant CARE 数据库对18个TadMTase基因上游2 000 bp 顺势作用调控元件分析,共预测出34种CREs顺式作用元件(图3A)。这些CREs除了2 种传统的启动子元件(TATA-box,CAAT-box)外,其余32种 CREs大致可以分为4组:植物生长发育、植物激素响应、光响应以及胁迫响应相关的作用元件(图3B)。所有已鉴定的小麦TadMTase基因启动子CREs位点共有460个,其中以光反应因子最多163个(35.43%),其次是激素反应因子144个(31.4%)、植物发育相关因子77个(16.74%)和胁迫反应因子76个(16.52%)。在TadMTase启动子中发现了10种光响应型CREs,其中,G-box 和sp1 CREs较多,普遍存在于TadMTase启动子,且拷贝数较多。参与激素信号转导的CREs包括生长素应答(TGA-box)、脱落酸反应(ABRE)、茉莉酸甲酯反应(CGTCA-motif 和 TGACG-motif)、赤霉素反应(GARE-motif 和 P-box)和水杨酸反应(TCA-element)元件。在这些CREs中ABRE、CGTCA、TGACG和TGA基序最为普遍。 TadMTase启动子还具有多种应激反应调控元件,如参与低温应激的 LTR,参与干旱诱导的MBS,涉及防御和应激反应的 TC-rich repeats以及响应厌氧诱导的 ARE 调控元件。另外,还确定了一些组织特异性的顺式调控元件,例如参与胚乳特异性表达所需的 GCN4_motif 和胚乳负调控相关的AACA_motif,以及与分生组织特异性激活有关的 CAT-box。此外,还发现参与玉米蛋白代谢调控的O2-site。总之,小麦TadMTase启动子中最常见的顺式调节元件主要与生理过程有关,例如光信号、激素响应和生长发育。 利用小麦基因组注释信息,对18个小麦TadMTase基因进行染色体定位。 结果表明,TadMTas分布于小麦21条染色体上的15条上(图4A),其中1A、1B和1D染色体上各有2个,其余3A、3B、3D,4A、4B、4D、5A、5B、5D、7A、7B、7D染色体各有1个TadMTase基因。在小麦A、B 和 D 同源染色体组中,均含有6个基因,说明TadMTase基因在小麦A、B 和 D 同源染色体进化过程中比较保守,不同基因在同源染色体的丢失和保留没有明显的偏好现象。利用MSCcanX软件对小麦全基因组产生的基因复制事件进行分析,结果发现18个TadMTase基因都存在与其高度相似的同源序列,并构成6个同源基因组,每组均由同源染色体组(A、B、D)上的同源基因组成,且这些同源基因组聚类分析均处于同一进化分支,如:TaDML3a-3A、TaDML3a-3B和TaDML3a-3D(图1)。同时分析了非同源染色体上的基因串联重复,其中在同一染色体上鉴定了3个基因串联重复,分别是TaROS1b-1A.1和TaROS1b-1A.2,TaROS1b-1B.1和TaROS1b-1B.2,TaROS1b-1D.1和TaROS1b-1D.2,而非同源的染色体上未发现基因串联重复(图4A)。 片段复制基因用彩色线条连接; 圆形代表AABBDD(Triticum aestivum)、菱形代表AA(Triticum urartu)、正方形代表DD(Aegilops tauschii)和 三角形代表AABB(Triticum dicoccoides)基因组中的dMTase 基因。 为进一步明确dMTase基因在小麦多倍体形成过程中基因扩张情况,利用TadMTase序列分别检索了AA(Triticum urartu)二倍体、DD(Aegilops tauschii)二倍体和 AABB(Triticum dicoccoides)四倍体小麦祖先种基因组。结果表明在祖先种的 AA、DD和AABB 基因组中分别检索到9、9和18个高度相似的dMTase同源基因,进化树显示TadMTase基因在其祖先物种中均有同源基因并在各自的进化分支,结果揭示在异源六倍体形成过程中存在部分dMTase基因丢失现象(图4B)。 为了进一步研究这些复制基因对受到何种选择,对小麦TadMTase基因Ks、Ka以及Ka/Ks比值计算。结果发现所有小麦TadMTase基因Ka/Ks<1,表明在基因扩张过程中经历了强烈的纯化选择,说明这些基因在进化过程中序列并未发生太大改变,相似性较高。 为了探讨不同生长发育阶段TadMTase基因在小麦中的组织特异性表达模式,利用小麦基因转录组数据库对 18个小麦TadMTase基因进行检索,分析了小麦花药(anth)、第1叶(lea_1)、胚(emb)、胚乳(en)、旗叶(fls)、颖片(glu)、籽粒(grain)、根(root)、茎(stem)和穗(spike)组织的转录水平,结果表明(图5A),小麦dMTase基因在不同组织或器官中的表达模式有明显的差异,TaROS1a-5A/5B/5D在所有组织中表达活跃,在穗和籽粒中表达量最高,而TaROS1b-1A.1、1B.1在胚组织中表达量较高,在穗和籽粒中也有表达。同时分析了TadMTase基因在籽粒不同发育时期(花后15、25和35 d)及其胚乳(en)和糊粉层(al)组织不同时期(花后10、20和30 d)表达情况,结果表明(图5B)小麦TadMTase基因的ROS1亚家族明显高于DML亚家族。其中TaROS1b-1A.1、TaROS1a-5A和TaROS1a-5D在籽粒、糊粉层及胚乳中表达量较高。在籽粒发育阶段,TaROS1b-1A.1、TaROS1a-5A和TaROS1a-5D表达量均在花后15 d最高,后逐渐下降。在糊粉层及胚乳发育阶段这3个基因表达量也均是前期高于后期。 小麦组织:花药、第1叶、胚、胚乳、旗叶、颖片、籽粒、根、茎和穗;dpa为籽粒、糊粉层、胚乳发育时期(花后天数)。下同。 基于小麦转录组数据中TaROS1a、TaROS1b和TaDML3亚家族基因在小麦籽粒发育过程中的表达量较高特征,继续在不同籽粒品质的小麦品种扬麦15(弱筋)和扬麦16(中强筋)进行了转录组测序分析,结果显示(图6),TaROS1a、TaROS1b和TaDML3亚家族基因在两种小麦籽粒发育中表达模式相似,但不同基因间表达量则发生了明显分化;TaROS1a-5A/B/D表达最为强烈,在弱筋小麦扬麦15中TaROS1a-5A/B/D表达在15 d达峰值后下降较缓慢,并持续至25 d后再迅速下降,而在扬麦16中TaROS1a-5A/B/D达峰值后则迅速下降,且TaROS1a-5A/B/D在弱筋小麦品种中表达量高于中强筋扬麦16;TaROS1b亚家族基因表达发生分化,TaROS1b-1A/B/D.1表达量较高,而TaROS1b-1A/B/D.2几乎不表达,且TaROS1b-1A/B/D.1在扬麦16中的表达则高于扬麦15;TaDML3表达量较低,且3个同源基因也发生分化,TaDML3-3A/B有少量表达,而TaDML3-3D则几乎不表达。 图6 TadMTase基因在扬麦15(A)和扬麦16(B)籽粒发育过程中的表达分析 基于TaROS1b-1A.1、TaROS1a-5A和TaROS1a-5D基因在小麦籽粒糊粉层和胚乳发育过程中的高表达量特征(图5B),为了进一步验证转录组的结果,根据获得的TaROS1b-1A.1、TaROS1a-5A和TaROS1a-5D序列设计引物(表1),用实时荧光定量 PCR方法分析3个基因在花后10、20和30 d籽粒种皮和胚乳组织种的表达情况。结果显示(图7)在种皮发育20 d时,TaROS1b-1A.1、TaROS1a-5A和TaROS1a-5D表达量均达最高,且TaROS1b-1A.1表达量高于TaROS1a-5A、TaROS1a-5D;在胚乳中,TaROS1b-1A.1和TaROS1a-5A两基因在20 d时表达量达最高,但TaROS1b-1A.1表达量低于TaROS1a-5A和TaROS1a-5D;在花后10 和20 d,弱筋小麦扬麦15籽粒种皮和胚乳中,3个基因表达量均显著高于中强筋小麦扬麦16。 字母代表同一时期不同品质小麦显著性差异比较(P<0.05)。 本研究从小麦全基因组数据库中获得了18 个 dMTase 基因成员,高于其它已报导物种中的基因家族成员数,结合系统发育分析,将TadMTases分为两类(ROS1和DML),其中DML又分为DML3、DML4和DML5。在小麦中没有发现DME亚族,这与其他研究发现的DME亚族基因只在拟南芥等双子叶中,而在单子叶植物中没有的结果一致[21-23]。基于双子叶植物中DME基因与ROS1基因的高同源性,推测DME基因是在单、双子叶植物分化后由双子叶植物内部ROS1基因复制产生的新基因[9]。 进化及保守结构域分析表明小麦dMTase与聚类关系较近的拟南芥和水稻dMTase具有相似的保守结构,如 ROS1 亚族的AtROS1、OsROS1a 和 TaROS1a-5A/B/D都含有 ENDO3c、FES、Perm-CXXC、RRM_DME 4个相同结构域。然而不同亚族之间保守结构域的个数和种类差异较大,这是可能是由于物种进化过程中产生基因无功能化、新功能化和亚功能化所致,说明dMTase基因在进化过程中会更加倾向趋异进化[24]。亚细胞定位研究还发现小麦dMTase均定位于细胞核,与其他植物物种,如拟南芥、水稻、番茄、花生、蓖麻豆、茄子、茶树和兰花也被证明位于细胞核内的结果一致。基于亚细胞定位在确定蛋白质功能中的重要性,植物dMTase基因的功能可能是保守的。 小麦dMTase在每个亚家族中具有相似的外显子-内含子组成,但ROS1和DML亚家族基因外显子-内含子数目变化有所差异,其中ROS1b亚家族基因的外显子-内含子数目为19~21个,而3个DML亚家族(DML3、DML4和DML5)基因均有相同的外显子-内含子,基因结构更为保守。 小麦dMTase基因上游 2 000 bp启动子序列的顺式作用元件分析发现,小麦dMTase的启动子具有光响应、植物激素响应、胁迫响应和生长发育 4 类CREs顺式作用调控元件,类似CREs已在其他植物如花生[25]、猕猴桃[26]、山茶花[18]和铁皮石斛[22]的dMTase基因启动子区也报道了ABRE、CGTCA-motif、TGA-element、G-box、sp1、A-box、ARE和CAT-box等元件,表明小麦dMTase基因可能参与了植物的发育和应激反应调控。其中TaROS1b-1D.2基因启动子中还发现了与种子特异性调控和胚乳特异性表达相关的CREs,表明TaROS1b-1D.2基因可能参与小麦胚乳发育的调控。 进化分析表明小麦dMTase基因在A、B和D部分同源基因组中分布均匀,进化一致。通过小麦与其祖先物种的dMTase基因进化分析发现,小麦dMTase与其祖先种在基因数量上存在差异,虽然基因总数可能在小麦异源六倍体形成进化过程中经历了3个二倍体祖先种的两轮自然加倍,使得基因组也经历了2次扩张,造成dMTase基因数目增加,但并非3倍增加,存在部分dMTase基因的丢失,其原因可能是由于小麦形成进化过程中染色体重组或基因功能冗余造成。共线分析表明小麦dMTase基因的ROS1b亚家族存在串联复制,产生新的功能和亚功能化基因,这为进一步研究不同DNA去甲基化酶基因的作用和机制提供了线索。 基因表达模式可以提供相关基因功能的重要信息,小麦dMTase在组织特异性表达模式分析中,发现TaROS1a亚家族基因在所有组织中都有较高的表达,在其他植物物种中也发现了类似的结果,如花生中的ROS2[25]和DoDML3在铁皮石斛[22]在所有组织中都有相对较高的表达。而TaROS1b亚家族基因表达存在基因分化和组织特异性,其中TaROS1b-1A.1基因在小麦胚和胚乳中有较高的表达,与其启动子胚乳特异性表达元件相一致,而该基因的串联复制基因TaROS1b-1A/B/D.2在各组织及发育时期几乎不表达,说明不同小麦dMTase基因在植物生长发育过程的在不同发育阶段可能具有独特的功能,也说明DNA 去甲基化酶基因在进化过程中会更加倾向趋异进化。 研究表明水稻OsROS1aknock-in突变后导致胚乳早期发育失败和胚发育不完全[12], 而OsROS1显性负突变导致糊粉层增厚,其调控机制为OsROS1基因的突变,可导致调控糊粉层发育相关的转录因子RISBZ1和RPBF超甲基化,抑制其基因表达,从而导致糊粉层细胞层数的增加[27]。小麦转录组数据分析表明小麦TaROS1a-5A/D和TaROS1b-1A.1在籽粒及其糊粉层和胚乳中高表达,这也被我们采用qRT-PCR方法证实(如TaROS1b-1A.1和TaROS1a-5A/D)。同时,qRT-PCR表达分析发现TaROS1b-1A.1和TaROS1a-5A/D在强筋和弱筋品质小麦中表达水平也存在一定的差异,推测TaROS1基因可能对小麦胚乳品质形成有一定影响。这些发现提示小麦ROS1基因可能在小麦胚乳和种皮糊粉层的发育发挥着和OsROS1同样的调控作用。然而,这些基因是如何调控胚乳和糊粉层的发育及品质形成,需要进一步开展基因功能等相关研究来加以验证。1.2 小麦 dMTase蛋白理化性质、结构域及保守motif预测分析
1.3 系统发育树的构建、基因结构分析
1.4 TadMTase基因染色体定位、共线性和基因复制事件分析
1.5 TadMTase基因组织表达分析
2 结果与分析
2.1 TadMTase基因家族成员的筛选及其蛋白理化性质分析
2.2 TadMTase基因家族蛋白系统进化树构建及保守结构域分析
2.3 TadMTase基因家族基因结构和启动子顺式作用元件分析
2.4 TadMTase基因染色体定位和基因复制分析
2.5 TadMTase家族基因表达分析


3 讨论
3.1 小麦dMTases 的蛋白质结构特征
3.2 小麦dMTase的基因结构特征及进化
3.3 小麦dMTase基因的表达与生长发育的关系
猜你喜欢
辽宁农业科学(2021年1期)2021-03-17
中国农业大学学报(2020年8期)2020-07-22
广州大学学报(自然科学版)(2019年1期)2019-05-07
小学阅读指南·高年级版(2016年9期)2016-10-31
天津科技大学学报(2016年1期)2016-02-28
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10
现代检验医学杂志(2015年2期)2015-02-06
现代检验医学杂志(2015年2期)2015-02-06
沈阳医学院学报(2014年4期)2014-12-27
遗传(2014年3期)2014-02-28
