
山东省依企业申请药品生产质量管理规范符合性检查质量风险分析
2023-11-11 13:29曹鸿雁赵杰柴发永张杰柏建学
药学研究 2023年10期
曹鸿雁,赵杰,柴发永,张杰,柏建学
(山东省食品药品审评查验中心,山东 济南 250014)
2019年12月1日起施行的《药品管理法》[1]取消了药品生产质量管理规范(以下简称GMP)认证,但GMP仍然是药监部门检查和企业从事药品生产质量管理的根本遵循。《药品管理法》《药品注册管理办法》《药品生产监督管理办法》《药品检查管理办法(试行)》等法律法规更新升级后,提出了药品生产质量管理规范(以下简称“药品GMP”)符合性检查概念,并提出了新的更高要求。本文通过研究分析2021和2022年度依企业申请开展的药品GMP符合性检查情况,梳理总结各类检查情形和分析检查发现的质量风险,并有针对性地提出对策与建议。
1 药品GMP符合性检查依据的法律法规
药品GMP符合性检查,是药品监督管理部门依据法规规章等有关规定对药品上市许可持有人(以下简称持有人)、药品生产企业实施药品生产质量管理规范情况开展的监督检查。药品GMP符合性检查分为依企业申请和依监管需要两种情形。
更新后的国家法律法规对药品GMP符合性检查情形进行了明确规定:《药品注册管理办法》[2]第四十七条、第四十八条、《药品生产监督管理办法》[3]第十六条、第五十二条以及有关实施公告第三条、《药品上市后变更管理办法(试行)》[4]第八条、《药品检查管理办法(试行)》[5]第三十四条等。
药品GMP符合性检查涉及的法律法规较多,检查情形较为复杂。为进一步加强药品GMP符合性检查工作,全国多个省局结合本省情况,先后出台了相关政策文件,指导药品GMP符合性检查工作的开展。其中山东省药监局印发了《山东省药品生产质量管理规范符合性检查工作程序》[6],分别对依企业申请和依监管需要开展药品GMP符合性检查进行了明确规定,并通过政策解读对依企业申请的药品GMP符合性检查情形进行了阐述。
2 山东省依企业申请开展的药品GMP符合性检查情况
2.1 依企业申请开展的药品GMP符合性检查总体情况从新法实施后,依企业申请开展的药品GMP符合性检查数据显示,2020年度开展61家次、2021年度开展135家次、2022年度开展140家次。从历年检查数据看,企业申报数量逐年增加,且情形复杂多样。
梳理2021和2022年度依企业申请开展的药品GMP符合性检查发现,2022年度和2021年度申报数量差别不大,但2022年原料药申报数量比2021年增加了67.6%,中药饮片减少了64%。2021和2022年度检查剂型/种类、家/次数详见表1。

表1 2021和2022年度检查剂型/种类、家/次数统计
2.2 依企业申请开展的药品GMP符合性检查办理情形新修订法律法规提出了系列药品GMP符合性检查的要求,通过研究分析发现,2021和2022年度的申报检查情形基本一致。按现行国家法规体系,原料药不得委托他人生产,所以从检查情形看,原料药不涉及委托生产,无菌制剂和非无菌制剂申报检查情形基本一致。从表2中可看出,生产场地变更占比最高,该类检查情形主要涉及自产增加委托、自产变更委托、委托变更自产或在原址新建、改扩建生产车间/生产线等情形。2021和2022年度依企业申请开展的药品GMP符合性检查情形见表2。

表2 2021和2022年度依企业申请开展的药品GMP符合性检查情形
3 依企业申请开展的药品GMP符合性检查发现的质量风险分析
GMP认证经过了多个发展过程,目前药品企业生产质量管理水平整体较好[7],依据药品GMP正文及附录内容对2021和2022年度的检查数据进行分类汇总分析发现,缺陷最多的章节都是集中在质量控制与质量保证、文件管理、设备方面,GMP附录涉及的缺陷项目也较多。2021和2022年度依企业申请开展的药品GMP符合性现场检查缺陷分类情况见图1。
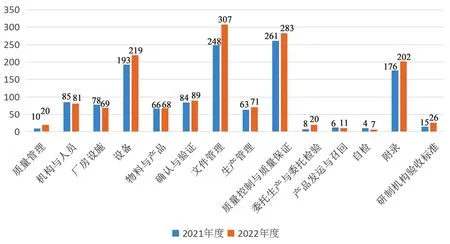
图1 2021和2022年度药品GMP符合性现场检查缺陷分类情况
从现场检查发现缺陷的风险程度看,不同企业、不同剂型的生产质量管理水平仍存在差异,其中在质量管理、生产管理、确认与验证等方面发现的问题比较典型,存在一定质量风险。
3.1 质量管理方面的问题质量风险主要集中在风险评估、持有人委托生产方面,典型问题有:①质量风险评估不到位:如个别企业未充分评估不同无菌类型产品共线生产可能存在的风险;②持有人委托管理不规范:持有人驻厂QA对委托生产的监督内容中缺少对生产管理、检验管理进行监督的具体规定;持有人对受托方现场审计、生产监督不规范;持有人与受托方部分公用文件内容规定不一致,可操作性不强,如变更、偏差、信息沟通等都有文件规定,但双方文件规定不一致。
质量风险管理是药品生产质量管理体系的重要组成部分,企业质量风险评估不充分或不到位,特别是尚未识别到的风险,会给产品质量带来安全隐患。随着药品上市许可持有人(MAH)制度的颁布实施,质量风险管理增加了新的内容,对MAH委托生产活动提出了新的挑战,在委托生产前、后,MAH应基于风险管理理念,根据科学知识及经验对受托企业开展全面有效的评估,特别是共线生产评估,确保将风险降低到可接受水平;另外,从检查数据分析看,MAH委托在现场审计、过程监督、文件衔接等方面也暴露出了问题,MAH应建立涵盖药品全生命周期的质量管理和文件管理体系[8],确保委托产品生产质量管理全过程受控。
3.2 生产管理方面的问题质量风险主要集中在生产管理不到位、防混淆防交叉污染能力方面,典型问题有:①无菌操作不规范:B级区操作人员走动步幅较大;分装操作人员多次进行扶倒瓶、剔除废瓶的干预操作,干预频率大于岗位操作规程规定次数;②显示已清洁的设备罐内存在部分白色膏状物残留;离心机相连物料管道三通处存在盲管,易残留物料,不易清洁;中药前处理提取车间集尘间的排风口已堵塞,未及时清洁维护;③洁净功能间个别中效、高效压差出现异常情况,未及时进行处理;④个别超过复验期的中药材已取样但未复检却用于投料生产。
产品是生产出来的,产品生产管理至关重要。从检查数据分析看,生产管理方面的风险,特别是无菌操作规范性方面的,会给产品质量带来风险。无菌制剂作为高风险产品,具有复杂性和特殊性[9],一直是检查的重点剂型。欧盟于2022年8月25日发布了新版《无菌药品的生产》GMP附录(2023年8月25日生效),对无菌药品生产和质量体系提出了新的更高要求,给无菌制剂生产企业带来了更大的挑战,企业应高度重视无菌制剂生产质量管理,科学评估无菌保障措施的有效性和适用性。
3.3 确认与验证方面的问题质量风险主要集中在确认与验证方案不完善、不全面,典型问题有:①企业未验证除菌过滤器重复使用次数;②原料药批量变更后企业进行了质量对比、杂质对比和1批稳定性考察,未进行工艺验证;③清洁验证擦拭取样点的选取不充分,未涵盖所有可能的风险点;④设备性能验证参数未涵盖实际使用参数;⑤培养基模拟灌装验证,未涵盖模拟传送轨道、自动进出料系统操作工位上的干预操作等。
确认与验证涉及面广,涵盖厂房设施与设备验证、工艺验证、清洁验证、分析方法验证、计算机化系统验证等方面[9]。从检查数据分析看,风险主要出现在设备验证、工艺验证、清洁验证方面,分析原因可能是评估不充分或考虑问题不全面,未合理确定验证的范围和程度,未科学评价验证产生的数据,导致相关工作做得不全面、不规范。
另外,在检查缺陷分析过程中发现,企业在数据管理方面也存在突出问题,质量风险主要集中在数据不完整、记录不及时、不便于追溯方面,典型问题有:①在原料药现场检查中发现数据真实性案例:动态生产工艺与《化药原料药生产工艺信息表》不一致,现场检查未通过;②气流粉碎机除尘滤网规定使用次数,但实际无证明使用次数的记录或台账;③灌装岗位人员剔除碎瓶的干预操作完成后未及时进行记录;④未记录退回物料的车间来源;⑤模拟灌装记录中未体现开门干扰和维修开门干扰的具体操作内容和时限。
《药品记录与数据管理要求(试行)》[10]明确了对记录与数据的基本要求,记录方面要求“保证全过程信息真实、准确、完整和可追溯”,数据方面要求“保证数据真实、准确、完整和可追溯”。记录与数据的管理贯穿药品生产质量管理的全过程,从检查数据分析看,企业在真实性、完整性、可追溯性方面存在一定风险,分析其原因可能是企业对记录与数据管理的认识或文件规定内容执行不到位,企业应高度重视记录与数据管理工作,科学评估数据可靠性管理以及数据在其生命周期内的每一个环节与期望之间的差距以及可能带来的数据管理风险[11],确保记录和数据真实、准确、完整、可追溯。
4 对策与建议
通过对山东省2021和2022年度依企业申请开展的药品GMP符合性检查情况分析发现,随着药品法律法规及政策红利的持续释放,企业申报的检查情形日趋多样化复杂化,为进一步做好依企业申请开展的药品GMP符合性检查工作,从企业和监管两个方面提出以下几点建议。
4.1 从企业角度①进一步落实持有人主体责任。MAH应当遵守《药品管理法》等相关法律法规,严格落实持有人主体责任,严格贯彻落实GMP及附录和不断更新的药品相关技术规范要求,不断健全完善药品质量管理体系,依法对药品生产全过程中药品的安全性、有效性、质量可控性负责;应强化风险管理,重点关注在生产质量管理方面存在的薄弱环节,加强对全生命周期的质量管理,特别是委托生产,要按照相关要求做好对受托方质量管理体系的审核和生产全过程的质量监督,做好委托方与受托方的体系衔接,保证产品质量;②进一步做好药品上市后变更管理工作。企业应加强对变更相关法规政策、技术指导原则的学习研究,与企业实际情况紧密结合,合理判断并准确厘清申请主体、申请范围、申请情形等,特别是变更生产场地,是上市后变更最复杂的情形,往往关联到生产工艺变更、工艺参数调整、生产设备更新、物料供应商变更、批量扩大等[12]。企业要确保经科学评估和必要的验证确定变更类别再实施变更,在贯彻实施GMP时落实好变更管理相关要求,有效预防合规风险。
4.2 从监管角度①精准把握药品法律法规政策。《药品管理法》实施后,药品相关法律法规、指导原则、技术指南等文件更新较为频繁,鉴于当前法规政策要求与企业申报情形的多样化、复杂性,需精准把握各药品相关法律法规的内涵及要求,科学高效开展现场检查;②明确药品GMP符合性检查重点。针对不同剂型、不同检查情形,明确检查重点[13-15],如,针对场地变更情形,应重点关注变更研究、变更评估及相关验证工作;针对原料药转“A”后上市前符合性检查事项,应在全面评估企业质量管理体系的前提下,重点关注企业现行工艺与已批准的化学药品工艺信息表的一致性,特别是基于风险未启动注册现场核查的品种。
总之,执行药品 GMP,是从事药品生产活动的基本要求。随着国家药品法律法规体系的不断完善更新,药品GMP符合性检查将在药品全生命周期发挥重要作用[7]。MAH应不断强化风险管理理念,落实好持有人主体责任;监管部门要不断增强识别风险的能力,督促持有人落实好主体责任,全面保障药品质量安全。
猜你喜欢
中外玩具制造(2022年4期)2022-04-08
中外玩具制造(2022年4期)2022-04-08
河北金融年鉴(2021年0期)2021-08-25
公民与法治(2020年12期)2020-07-25
公民与法治(2020年4期)2020-05-30
中国宝玉石(2017年6期)2018-01-13
公民与法治(2016年9期)2016-05-17
华东师范大学学报(自然科学版)(2014年3期)2014-03-11
机电信息(2014年35期)2014-02-27
机电信息(2014年20期)2014-02-27
