
Co 掺杂ZnO/SiO2 复合脱硫剂常温脱硫性能及再生性能
2023-11-29 12:52苏哲林李敏麒王业双樊惠玲
煤炭学报 2023年10期
苏哲林,李敏麒,杨 超,王业双,樊惠玲
(1.太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室,山西 太原 030024;2.太原理工大学 环境科学与工程学院,山西 太原 030024)
煤炭在我国能源结构中占主导地位,但在其清洁高效利用过程中不可避免的会产生含硫化合物,其中以无机硫H2S 为主。后者不仅会毒害下游催化剂,腐蚀管路和设备[1-2],而且污染环境,危害人体健康。金属氧化物和硫之间存在较强的相互作用,普遍用于H2S 的脱除。由于ZnO 脱硫剂[3-4]脱硫精度高,在工业应用中通常作为精脱硫的最后一道工序。从热力学角度,其在常低温条件下有更好的脱硫精度。但受反应动力学的制约,传统ZnO 脱硫剂在常温条件下的脱硫活性非常差。
金属掺杂是提高ZnO 常温脱硫性能的有效策略。BEZVERKHYY 等[5]发现Cu+掺杂ZnO 在脱硫过程中由于“电荷补偿效应”增加了ZnS 中硫空位的密度,加快了S2-与O2-的交换速率,从而提升了ZnO 的脱硫性能。YANG 等[6]发现Ni2+掺杂ZnO 可增加其氧空位的密度,促进ZnO 表面羟基化以及HS-/S2-与O2-的交换速率。Co2+也常被用作掺杂剂用以调变ZnO的脱硫性能。BAIRD 等[7]采用浸渍法和共沉淀法制备Co 掺杂的ZnO 脱硫剂,穿透实验研究表明,Co 掺杂可提高ZnO 的分散性,增加ZnO 的比表面积,有利于提高ZnO 对H2S 的脱除能力。但YANG 等[8]在研究不同金属离子掺杂对ZnO 常温脱硫性能的影响时,发现Co 掺杂对ZnO 脱硫剂的脱硫性能无明显影响。上述结果表明,Co 掺杂对ZnO 脱硫性能的影响规律仍存在争议,需进一步研究。
目前,中低温金属氧化物脱硫剂在使用后很难再生循环利用,因此会形成大量固废。近年来,国家环保法规的日益严苛,企业固废处理面临很大的压力。因此,脱硫剂的再生循环性能仍是制约其利用的瓶颈。在含氧气氛下高温加热是ZnO 脱硫剂常用的再生方法,但该过程易导致ZnO 纳米颗粒团聚和硫酸盐的生成[9],影响再生效率。在脱硫剂中掺杂过渡金属可改善其再生性能,但掺杂不同的金属离子对其影响不同。研究表明Cu2+掺杂可降低ZnO 脱硫剂的再生温度[8]。而Ni2+掺杂不仅使ZnS 再生过程中生成硫酸盐,导致再生不完全,而且会造成再生过程中结构部分坍塌[10]。虽然Co 掺杂常用于改善ZnO 脱硫剂的脱硫性能,但目前关于Co 掺杂ZnO 常温脱硫剂再生性能的研究却鲜有报道。
笔者制备了掺杂不同量的CoZnO/SiO2复合脱硫剂,考察并分析Co 掺杂对ZnO 常温脱硫性能的影响规律,同时考察Co 掺杂ZnO 脱硫剂的再生性能,并分析影响脱硫剂再生性能的主要因素。
1 实 验
1.1 脱硫剂的制备
取正硅酸乙酯、去离子水、稀HNO3、无水乙醇,按照摩尔比为1∶1.8∶0.03∶3.9 配制混合溶液,磁力搅拌1 h 得到SiO2溶胶;将适量硝酸锌和硝酸钴加入到甲醇和乙二醇的混合溶剂中,其中乙二醇和甲醇的体积比为3∶2。将完全溶解的硝酸盐溶液加入到SiO2溶胶中并继续磁力搅拌2 h,得到混合溶胶,其中(Co+Zn)/Si 的摩尔比为1∶1。将上述混合溶胶置于真空烘箱中30 ℃老化10 h,再升温至120 ℃干燥8 h;将干燥后的凝胶转移至马弗炉中,程序升温阶段(升温速率为1 ℃/min)在300 ℃下焙烧2 h 后,升温至500 ℃焙烧2 h 得到脱硫剂。脱硫剂命名为ZCX(X为Co 掺杂的摩尔分数),吸附脱硫后的脱硫剂命名为ZC-XS,再生后的脱硫剂命名为ZC-XR,多次再生后的样品命名为ZC-XR-Y(Y为再生次数);以商业氧化锌脱硫剂作为参考,命名为ZnO-C,再生后的样品命名为ZnO-CR。
1.2 表征方法
采用D/max-2 500 型X 射线衍射仪对脱硫剂的物相进行分析;采用超高分辨率场发射扫描电镜(TESCAN MAIA 3LMH)对脱硫剂的表面形貌进行观察;脱硫剂的微观形貌采用场发射电子显微镜(Tecnai G2 F20)观察和分析;采用物理吸附仪(Micromeritics ASAP 20 200 PLUS HD88 型全自动物理吸附仪,美国麦克仪器公司)表征脱硫剂再生前后的比表面积和孔结构。采用Brunaure-Emmett-Teller(BET)方法计算脱硫剂的比表面积,采用Barrett-Joyner-Halenda(BJH)方法计算脱硫剂的孔径分布;采用X 射线光电子能谱仪(ESCALAB 250)分析脱硫剂脱硫前后的表面电子结构变化。
采用化学吸附仪(Aurochem II-2920)分析脱硫剂的表面酸碱性质,实验方法:称取样品0.1 g,在200 ℃下通入He 气吹扫1 h,气体流速为50 mL/min,自然降温至室温后,采用CO2在70 ℃下处理样品30 min,气体流速为30 mL/min,再He 气吹扫1 h,将温度从室温升至600 ℃,升温速率10 ℃/min,实验使用TCD 检测CO2信号。
1.3 脱硫剂脱硫和再生性能测试
脱硫剂的脱硫和再生性能测试均在U 型管固定床反应器中进行,反应器内径为6 mm。将制备的样品研磨筛分,选择颗粒尺寸为0.250~0.425 mm(40~60 目)的样品,将其装填到反应器中,样品装填高度为2 cm。脱硫实验开始前,采用湿N2对样品吹扫2 h,再将N2稀释后的H2S 气体通过水饱和器后通入到U 型反应器中,脱硫反应温度为30 ℃,进口H2S的质量浓度为(1 400 ±0.05)mg/m3,气体流速为100 mL/min。当出口H2S 质量浓度达到进口的0.1%时视为穿透;当进出口气体中H2S 质量浓度相同时,样品达到吸附饱和,停止实验。采用微量硫分析仪对反应器进出口H2S 质量浓度进行检测分析,脱硫剂的穿透硫容和饱和硫容通过积分脱硫剂的穿透曲线计算。
脱硫剂的再生实验通过程序升温方式进行。实验前,使用N2吹扫饱和后的脱硫剂1 h,以去除物理吸附和管路中残余的H2S。然后采用程序升温将再生温度由室温升至800 ℃,恒温至出口SO2质量浓度低于检出限,停止再生实验。该过程中升温速率为1 ℃/min,再生气氛为N2+O2,其中O2体积分数为3%,气体流速为100 mL/min。采用微量硫分析仪对反应器出口的SO2质量浓度进行检测。再生尾气经KOH 吸收液后排空。
2 结果与讨论
2.1 新鲜样品表征
图1(a)为所制备脱硫剂的XRD 谱图。由图1(a)可知,所有样品中均出现了纤锌矿ZnO 的特征衍射峰,但其衍射峰的强度较弱,表明所制备脱硫剂中ZnO 晶粒较小,分散较好。在脱硫剂中引入Co 物种后,ZnO特征衍射峰的强度进一步减弱。可见Co 物种的引入有利于ZnO 晶粒尺寸的减小,这可能是由于引入的Co 物种掺杂到ZnO 晶格中,抑制了ZnO 晶粒的生长[11]。XRD 谱图中未观察到Co 物种的特征衍射峰,说明Co 物种可能掺杂到ZnO 晶格中,但也不能排除Co 物种的晶粒尺寸非常小,超出了XRD 检测仪器的检测下限。
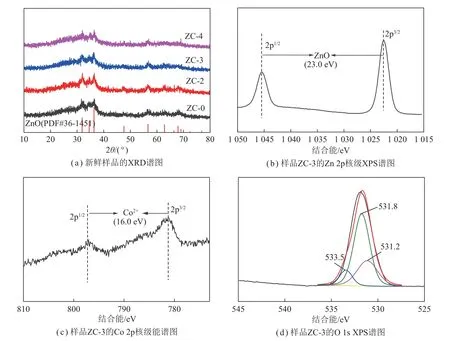
图1 新鲜样品的XRD 谱图和样品ZC-3 高分辨Zn 2p、Co 2p 和O 1s XPS 谱图Fig.1 XRD patterns of fresh samples and XPS spectra of Zn 2p,Co 2p and O 1s of ZC-3
为了进一步明确Co 物种在脱硫剂中的存在形态,选取ZC-3 进行XPS 表征。图1(b)为该样品的Zn 2p核级XPS 谱图,Zn 2p 裂分为Zn 2p3/2和Zn 2p1/2轨道,两者能差为23.0 eV,表明脱硫剂中存在ZnO[12]。图1(c)为该样品的Co 2p 核级能谱图,Co 2p3/2和Co 2p1/2轨道结合能分别为781.3、797.3 eV,两者能差为16.0 eV,表明Co 物种在ZnO 脱硫剂中的存在形式为Co2+[13]。图1(d)为该样品的O 1s XPS 谱图,经高斯分峰发现脱硫剂中O 的存在形式主要有3 种,即晶格氧 (531.2 eV)、表面羟基氧 (531.8 eV)[6]和表面吸附氧 (533.5 eV)[14],其中,表面羟基氧占总氧的48.1%,说明Co 掺杂的ZnO 中存在较多的晶格缺陷。
图2 为ZC-3 的SEM 和TEM 图片。由图2 (a)可见,新鲜脱硫剂是由许多纳米颗粒密堆积而成,纳米颗粒之间存在许多堆积孔。TEM 结果表明,ZnO纳米颗粒尺寸分布在20~30 nm(图2(b))。高分辨TEM 结果(图2(c))表明,脱硫剂中ZnO 纳米颗粒暴露的晶面主要为(101)和(100)面,未发现与Co 物种有关的晶格条纹,结合XRD 和XPS 结果说明引入的Co 物种成功掺杂到ZnO 晶格中,并非以单独的物相存在。

图2 ZC-3 的SEM、TEM 图片Fig.2 SEM and TEM image of ZC-3
脱硫剂的织构性质如图3 所示。图3(a)表明,所有脱硫剂的吸附曲线均属于Ⅳ型吸附曲线,且具有明显的H2 型滞后环,说明所制备的脱硫剂均为介孔材料。引入Co 物种后,脱硫剂对N2的吸附量明显增加,比表面积和孔容增大(表1),但Co 物种的加入量对两者的影响并不显著。由图3(b)可见,氧化锌中掺入Co 物种,脱硫剂的平均孔径由原来的8.0 nm 增到12.5 nm。脱硫剂的孔隙结构主要来源于纳米颗粒堆积和SiO2基体(图2),引入Co 物种后脱硫剂织构参数的变化可能是由于Co 物种的引入使ZnO 晶粒变小、分散性提高所致。
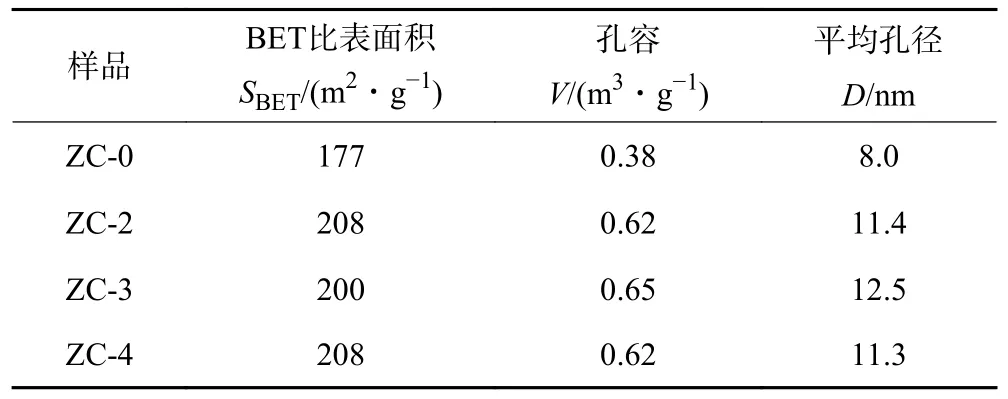
表1 新鲜脱硫剂的织构参数Table 1 Texture parameters of fresh desulfurizers
脱硫剂表面酸碱性采用CO2-TPD 进行表征,结果如图4 所示。在100~250、250~400 和400~500 ℃三个温度区间,脱硫剂均出现了CO2脱附峰,分别对应脱硫剂表面存在的弱碱性位点羟基官能团、中强碱性位点Zn-O 离子对[6]、强碱性位点(即表面低配位的O2-物种[15])。Co 物种掺杂导致脱硫剂表面碱性位点显著增强,由XPS 结果可推测,这可能与Co 掺杂使ZnO 表面晶格缺陷增多有关。
2.2 样品的脱硫性能及分析
图5 为本文制备的脱硫剂和商业ZnO 脱硫剂的穿透曲线及硫容。由图5 可知,ZC-0 的穿透硫容和饱和硫容分别为91 mg/g 和107 mg/g,ZnO 的利用率为47.1%。掺杂Co 物种后,脱硫剂的脱硫性能和活性组分利用率明显提高,当Co 的掺杂量为3%时脱硫剂的脱硫性能最高,穿透硫容和饱和硫容分别为143 mg/g和165 mg/g,经估算ZnO 的利用率至少在71.8%以上。商业ZnO 脱硫剂ZnO-C 的穿透硫容和饱和硫容分别为42.8 mg/g 和44.1 mg/g,明显低于本文制备的脱硫剂。
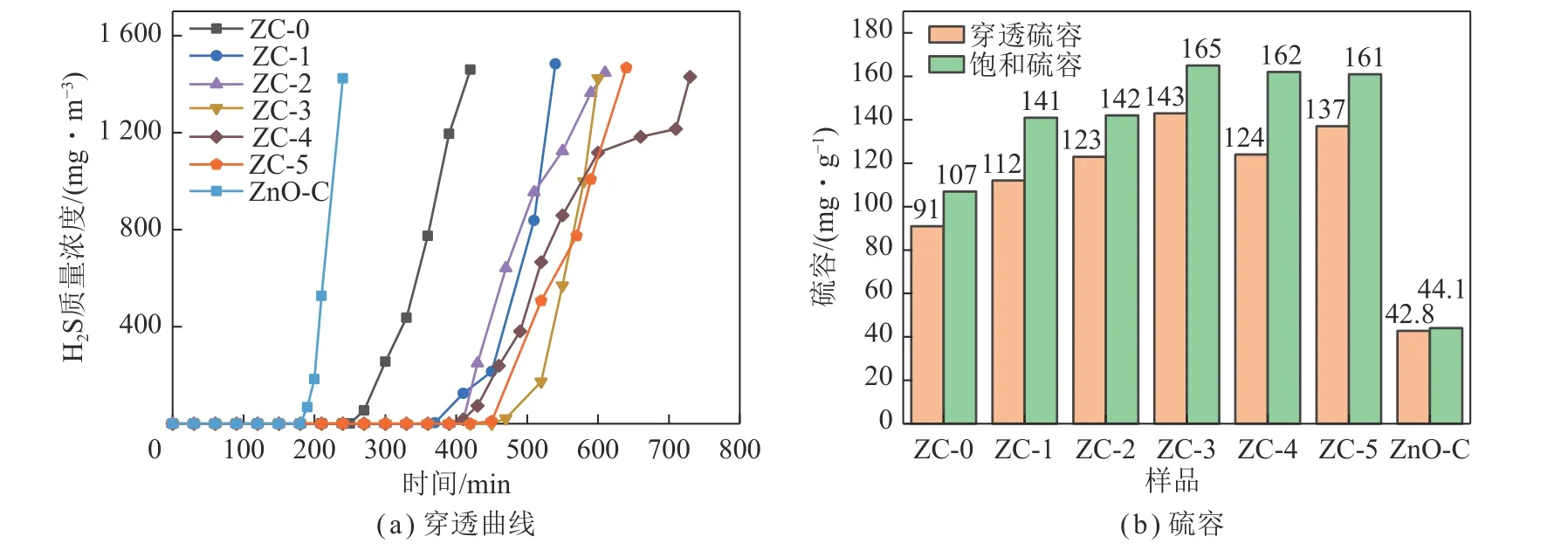
图5 不同脱硫剂的穿透曲线及硫容Fig.5 Breakthrough curves and sulfur capacity of desulfurizers
对部分硫化样进行XRD 与XPS 表征。图6(a)为硫化样的XRD 谱图。除了ZC-0 的硫化样品中仍可见非常微弱的ZnO 特征衍射峰外,其余硫化样中ZnO 的特征衍射峰几乎消失。各样品中可见归属于ZnS 的较弥散特征衍射峰,未见Co 的硫化物种,说明脱硫过程中ZnO 与H2S 发生化学反应生成ZnS,且掺杂Co 的脱硫剂反应程度更高。图6(b)为硫化样ZC-3S 的S 2p 核级能谱图。在结合能为161.6、162.8 eV处出现2 个拟合峰,前者对应ZnS 或CoS 物种,后者归属为CoSOH 物种[16]。为进一步证实硫化样中Co的存在状态,对硫化样ZC-3S 中的Co 物种进行XPS分析,如图6(c)所示。Co 2p3/2轨道在结合能780.2、783.3 eV 处出现2 个拟合峰,分别归属于CoSOH[17]和CoS[18]。由于本实验的进料气中无氧,所以推测CoSOH 可能是由于在表征过程中,硫化物中的CoS与空气接触发生氧化所致[19]。
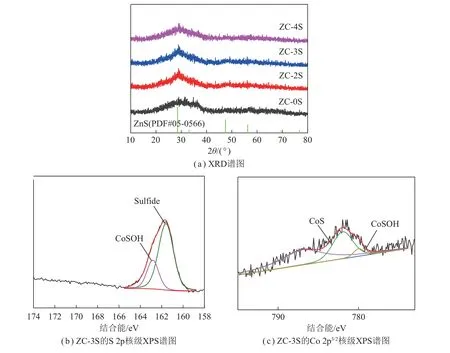
图6 硫化样的XRD 谱图、硫化样ZC-3S 的S 2p 和Co 2p 核级XPS 谱图Fig.6 XRD patterns of vulcanized samples,and XPS spectra S 2p and Co 2p of ZC-3S
脱硫剂的常温脱硫性能通常与脱硫剂活性组分粒径、比表面积和孔结构有关。晶粒越小、比表面积越大,越有利于暴露更多的活性位点;大孔径则有利于脱硫过程中的气体传质。Co 物种引入后,脱硫剂的晶粒变小、比表面积明显增加,而且孔径变大,使得ZnO 脱硫性能提高。另外,由XPS 与CO2-TPD 结果可知,Co 物种引入后,ZnO 表面碱性增强,为脱硫过程中H2S 的解离提供有利环境。同时,由于ZnO 与H2S 的反应为气固非催化反应,反应过程涉及硫氧离子交换,Co 掺杂所引起的晶格缺陷可促进S2-在ZnO晶格中的固体离子扩散,从而提高ZnO 的脱硫性能。
2.3 脱硫剂再生性能及分析
为了探究Co 掺杂对ZnO 脱硫剂再生性能的影响,选取ZC-0S 和ZC-3S 进行程序升温氧化实验。由于在氧化温度低于300 ℃时,2 个样品几乎检测不到SO2的释放,故实验结果从300 ℃起呈现。由图7 可见,2 个硫化样均在550、710 ℃出现SO2的强释放峰,前者对应硫化样中ZnS 氧化生成ZnO 与SO2过程,后者由硫酸锌的分解所致,即硫酸锌分解为ZnO 和SO2[20]。脱硫剂的脱硫产物为ZnS 和少量CoS,并未有硫酸盐生成,因此推测硫酸盐是由于在程序升温氧化过程中ZnS 过度氧化,或再生后的ZnO 继续与生成的SO2发生反应导致。对比ZC-0S 和ZC-3S 的再生曲线发现,Co 掺杂的硫化样品释放的SO2体积分数明显高于未掺杂样品,这与穿透实验的结果吻合。另外,与ZC-0S 相比,ZC-3S 由硫酸盐分解产生的SO2体积分数显著降低,而ZnS 直接氧化产生的SO2体积分数显著增加。说明Co 掺杂能显著抑制ZnO在再生过程中形成的硫酸盐量。抑制再生过程中硫酸盐的生成对脱硫剂的再生性能提高有重要意义。金属氧化物脱硫剂的氧化再生大部分选择600 ℃以上的高温,通常也是考虑到分解硫酸盐,使再生完全。若能减少硫酸盐的生成,再生温度可降低,不仅利于节能环保,也能使脱硫剂免于因高温烧结引起的结构损坏,进而导致脱硫剂的循环性能下降。从图7 可见,要使脱硫剂再生完全,ZC-0S 的再生温度明显高于ZC-3S。
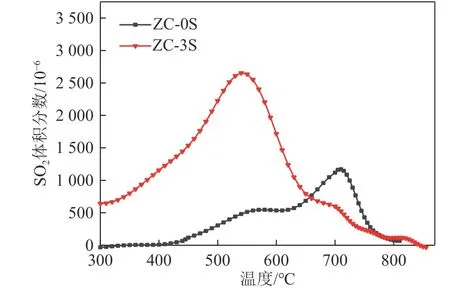
图7 脱硫剂再生过程中SO2 出口体积分数随温度的变化Fig.7 Relationship between SO2 outlet concentration and temperature in the process of desulfurizers regeneration
为进一步明确Co 物种掺杂对ZnO 脱硫剂再生性能的影响,选取ZC-0、ZC-3 样品在650 ℃进行脱硫再生。另外,为了说明所制备脱硫剂的再生性能,对商业ZnO 脱硫剂也进行再生评价。再生后脱硫剂的脱硫性能如图8 所示。相比于新鲜样品,再生后脱硫剂的脱硫性能均明显下降。但ZC-3 脱硫剂的再生性能明显高于ZC-0,其再生3 次后的脱硫性能仍与新鲜的商业ZnO 脱硫剂的饱和硫容相当,说明Co 掺杂对ZnO 脱硫剂再生性能具有促进作用。
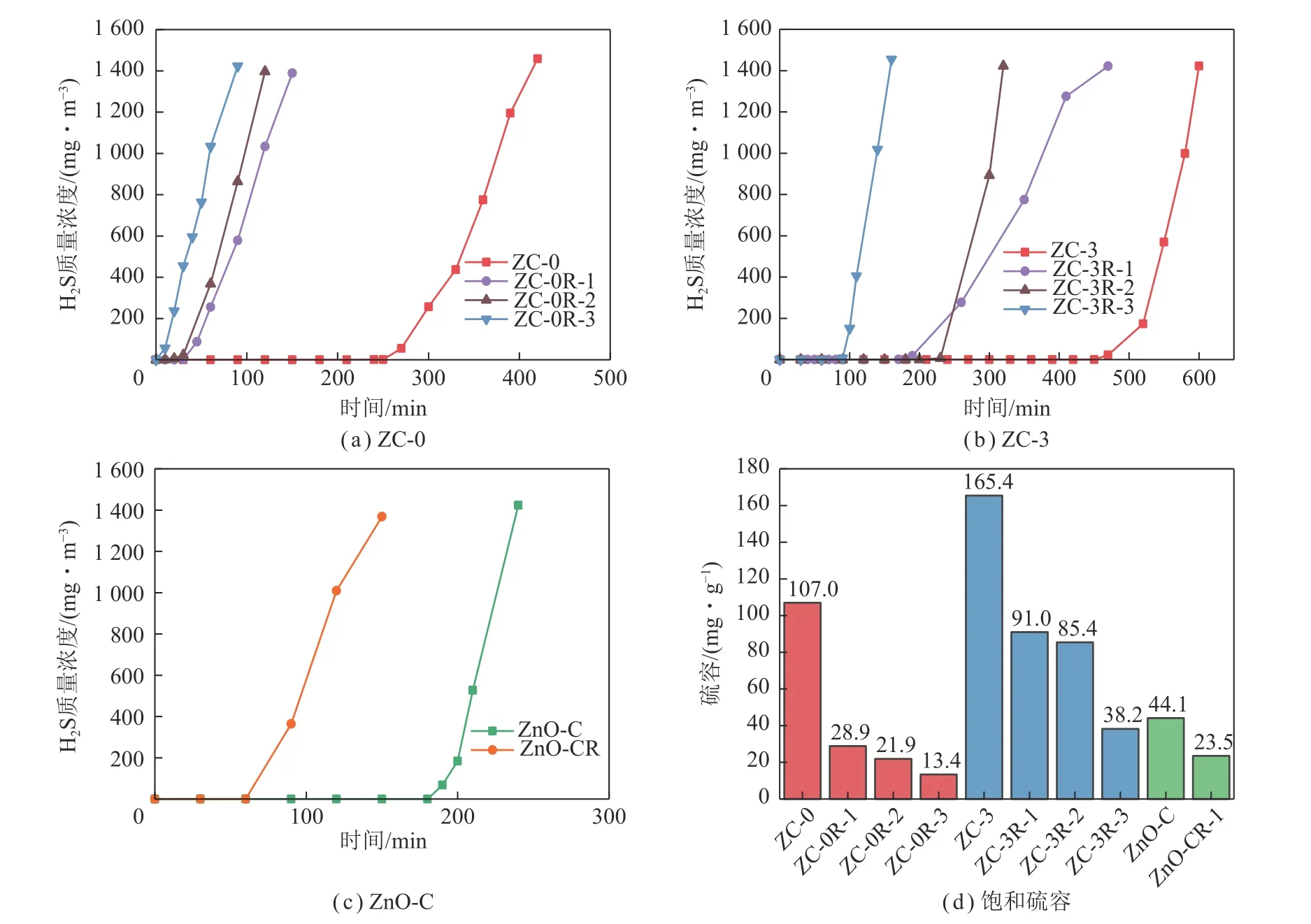
图8 脱硫剂ZC-0、ZC-3 和ZnO-C 的脱硫/再生循环实验的穿透曲线和饱和硫容Fig.8 Breakthrough curves of ZC-0 and ZC-3 and ZnO-C in desulfurization/regeneration cycle experiments and saturated sulfur capacity
为阐明样品ZC-3 再生性能下降的原因,选取第1 次再生后的样品ZC-3R-1 进行表征,结果如图9 所示。由图9(a)可知,再生后的样品中不仅出现ZnO的特征衍射峰,还出现了ZnSO4的特征衍射峰,说明样品在再生过程中并未完全再生。由图9(b)可知,再生后脱硫剂的比表面积和孔体积显著下降(表2),孔径分布如图9(c)所示。再生后脱硫剂的孔径由单孔径转变为双孔径,平均孔径由12.5 nm 降至8.1 nm,这可能与硫酸盐的生成有关。同时由于再生是强放热反应,局部温度过高也有可能导致脱硫剂的孔隙结构坍塌或ZnO 纳米颗粒团聚。图9(d)为再生样的CO2-TPD 曲线,与新鲜样相比,再生后样品的表面碱性显著下降,这与再生后脱硫剂中硫酸盐的存在相关。另外,再生后ZnO 纳米颗粒团聚,表面晶格缺陷减少也可能是导致脱硫剂碱性下降的原因。

表2 脱硫剂再生前后的织构参数Table 2 Texture parameters of samples ZC-3 and ZC-3R-1

图9 新鲜样品ZC-3 和再生后样品ZC-3R-1 的XRD 谱图、吸附等温线、孔径分布和CO2-TPDFig.9 XRD patterns,adsorption isotherms,pore size distribution and CO2-TPD of fresh sample ZC-3 and regenerated sample ZC-3R-1
结合表征结果可知,虽然可通过Co 掺杂抑制氧化锌脱硫剂在再生过程中形成硫酸盐,但不能完全避免。且由于再生是强放热过程,脱硫剂在较低温度下再生,比表面积仍会显著下降。这些因素使得再生后脱硫剂的工作硫容明显下降。获得可循环利用的常温氧化锌脱硫剂仍存在非常大的挑战,通过控制再生速率或通过孔隙改善来缓解再生局部放热,以保持脱硫剂结构的稳定或许有助于进一步提高氧化锌脱硫剂的再生性能,将是后续研究的内容。
3 结论
(1)Co 掺杂不仅可增大ZnO 脱硫剂的比表面积和孔容,还可提高ZnO 晶格缺陷量,促进表面羟基化,进而增加脱硫剂表面碱性。
(2)Co 掺杂显著提高了ZnO 脱硫剂的脱硫性能,当Co 掺杂量为3%时,ZnO 脱硫剂的脱硫效果最佳,穿透硫容高达143 mg/g,约为ZnO 脱硫剂的1.6 倍。
(3)Co 掺杂可显著抑制ZnO 脱硫剂在再生过程形成硫酸盐,但并不能完全避免硫酸盐的生成。
(4)Co 掺杂ZnO 脱硫剂再生性能下降的主要原因是由于再生过程中形成硫酸盐且不易再生完全,ZnO 脱硫剂在较低温度下的氧化再生过程中仍会发生明显的结构损失。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
辽宁化工(2022年6期)2022-07-01
工程技术与管理(2022年2期)2022-03-04
云南化工(2021年5期)2021-12-21
装备维修技术(2020年4期)2020-11-23
冶金动力(2020年5期)2020-06-15
四川冶金(2019年5期)2019-12-23
山东冶金(2019年2期)2019-05-11
经济技术协作信息(2018年30期)2018-11-22
安全、健康和环境(2018年7期)2018-08-03
