
偶氮四唑钾盐的绿色电合成反应耦合WS2纳米片催化电解水制氢
2023-12-19 14:38姚田浩马语和柳博龙马玉强李嘉辰马海霞
高等学校化学学报 2023年12期
姚田浩,马语和,柳博龙,马玉强,张 聪,李嘉辰,马海霞
(西安市特种能源材料重点实验室,西北大学化工学院,西安 710069)
迫于能源快速消耗和环境污染的压力,氢(H2)作为高能量密度的清洁能源载体,有望取代日益枯竭的化石燃料[1].电解水析氢是一种理想的制氢方法[2].电解水包含析氢反应(HER)和析氧反应(OER)两个半反应.
目前,最有效的HER 催化剂是贵金属(如Pt 和Pd)基,但其稀缺性和高昂的成本阻碍了其大规模应用[3].近年来,成本低、储量大、具有优异HER催化性能的层状过渡金属二硫化物(TMDs)被广泛研究[4].而TMDs 中的二硫化钨(WS2)因活性高、化学稳定性好、资源丰富、层状结构活性位点及类似于自然界中生物素酶的特点促进Heyrovsky/Tafel步骤氢中间体(H*)的形成而被认为是一种具有发展前景的HER催化剂候选材料[5,6].
目前,关于HER催化剂的研究主要围绕单原子催化剂(Ru单原子、Pt单原子)、过渡金属基催化剂(硫化物、磷化物等)和非金属催化剂(碳纳米管、石墨烯等)而展开[7],并通过构建不同维度的结构形貌[如一维(纳米线等)、二维(纳米片等)、三维(3D纳米管等)]使催化剂暴露更多的活性位点以及提高比表面积从而使催化性能得到提升[8],其中,二维纳米材料因具备高比表面积及高密度的缺陷和边缘,目前,已被广泛用作提升HER催化剂催化性能的策略[9].WS2作为一种半导体二维材料,其HER活性因其低导电性、边缘活性位点较少的特点而受到限制[10].因此,通过引入良好导电基底(如碳布),并在其表面生长WS2纳米片(NSs),在暴露出更多的活性位点、增大材料接触比表面积的同时增强了导电性,提高了电子的传输效率,从而提高电催化性能[11~14].
OER是动力学速率缓慢的四电子多步骤过程,由于其较高的理论氧化电位导致了电解水过程中高的能耗,被认为是电解水过程的瓶颈反应,且OER过程产出的氧气(O2)经济价值较低,与氢气混合可能会导致爆炸,因而用具有热力学较低氧化电位的阳极反应取代OER,从而使全解水槽压更低的同时获得经济价值更高的化工产品的策略引起了广泛关注.近年来,利用乙醇[15]、甘油[16]、甲醇[17]、尿素[18]、5-羟甲基糠醛[19]和水合肼[20]等有机小分子的氧化反应代替OER,以实现全解水的超低电解电压与生产高附加值产品成为研究热点.
有机分子5-氨基-1H-四唑(5-AT)因其氮含量高、热稳定性好,近年来,备受含能材料领域研究的关注,作为一种常用的含能材料配合物,5-AT是制备5-硝基四唑汞以及双5-氨基四唑铜的基本原料,可通过氨基胍重氮异构法、叠氮胍硝酸盐法和什托列法等合成[21,22].以5-AT 为原料,可得氧化产物5,5'-偶氮四唑二钾盐(K2AZT),K2AZT作为一种含能材料具备高能量密度、能够储存大量化学能,并在一定外界刺激下能够快速分解瞬间释放出巨大能量的特点,含能材料目前广泛用于军事燃烧及爆破、火箭与导弹运载、推进剂及消焰剂等领域[23].通过传统化学合成反应制备K2AZT 需要加入强氧化剂KMnO4,且反应过程伴随着副反应,纯化过程复杂[24,25].将K2AZT 的绿色电化学合成与HER 偶联构成耦合体系的方式,可以避免传统氧化方式的不足,且具有易于放大的特点和低成本合成等优点[26].同时,研究发现某些偶氮反应的电合成相比于OER具有更低的氧化电位[27].因此,用热力学较低氧化电位的5-AT的氧化偶联取代OER并与HER耦合,能够在低电压下产生高纯氢气,同时安全、方便、绿色地将5-AT氧化偶联成K2AZT.
1967 年,Wawzonek 等[28]在非水介质中以Pt 作为阳极,以电化学的方式初次实现了N=N 键的形成.1998 年,Hiskey等[29]以KMnO4作为氧化剂,在碱性水溶液中将5-AT氧化偶联合成了K2AZT.2018年,He 等[30]以BiVO4薄膜作为光电阳极,在室温下实现5-AT 的氧化偶联合成偶氮四唑盐.2020 年,Fu等[25]首次探究了在碱性室温条件下多种阳极电极以电化学方式驱动5-AT的氧化偶联的可行性,初步发现包含Pt,Au,Ag等贵金属以及包含Cu,Ni,Co,Mo在内的过渡金属作为阳极金属具备将5-AT氧化偶联为偶氮四唑盐的能力,但对其催化能力大小没有进行横向的对比.Pt,Au,Ag等贵金属存在商业化使用成本过高的问题,而过渡金属Ni,Mo地球储量不丰富.而在Cu,Ni,Ti几种金属中,Cu本身具有优越的易得性、易于进行放大实验、导电性好的特点,3D结构的泡沫铜(CF,Copper foam)具有更大的相对表面积,并且提供了更多的电荷载体转移途径,因而具备更优越的催化活性,除此之外,3D结构也提供了抵抗机械变形的能力[31].因而,选用了泡沫铜(CF)、泡沫镍(Ni)和钛片(Ti)用以探究最佳电极.
本文建立了一种新的耦合体系,在阴极以CC@WS2NSs作为HER催化剂发生析氢反应的同时,阳极以5-AT氧化偶联反应取代OER反应,通过绿色电化学合成了含能材料.
1 实验部分
1.1 试剂与仪器
碳布[CC,厚(0.36±0.02)mm],上海三马斯克有限公司;泡沫铜(CF,厚0.3 mm),昆山广佳源新材料有限公司;氢氧化钾(KOH,纯度≥85.0%)、5-氨基-1H-四唑(CH3N5,5-AT,纯度≥98.0%)和二水合钨酸钠(Na2WO4·2H2O,纯度99.5%,分析纯),上海阿拉丁生化科技有限公司;二水合乙二酸(C2H2O2·2H2O,纯度≥99.5%,分析纯),国药集团化学试剂有限公司;硫代乙酰胺(C2H5NS,纯度≥99.5%,分析纯),成都科隆化学品有限公司;H 型电解槽的阴离子交换膜(AEM,Fumasep FAA3-50),德国Fuma-tech公司.
CHI760E 电化学工作站,上海辰华仪器有限公司;Bruker D8 Advance X 射线粉末衍射仪(XRD)和AVANCE 500MHz 核磁共振波谱仪(NMR),德国Bruker 公司;SU8010扫描电子显微镜(SEM),日本Hitachi 公司;Talos F200X 透射电子显微镜(TEM)和Thermo Scientific Nexsa X 射线光电子能谱仪(XPS),美国Thermo Fisher Scientific 公司;IRAffinity-1S傅里叶变换红外光谱仪(FTIR),日本Shimadzu公司;Optima 2100DV全谱直读等离子体发射光谱仪,美国PerkinElmer公司;Q2000/Q600-TA差示扫描量热仪/同步热分析仪,美国TA公司.
1.2 实验过程
1.2.1 CC@WS2纳米片的制备 将裁剪好的CC(1 cm×3 cm)浸泡在乙醇溶液中,超声清洗10 min后,用去离子水和无水乙醇依次清洗数次,以去除CC 表面杂质.将0.9 mmol Na2WO4·2H2O 溶解于15 mL 去离子水中,待完全溶解后,加入5.25 mmol C2H5NS,充分溶解后加入1.67 mmol草酸(C2H2O4·2H2O),搅拌10 min完全溶解得乳白色溶液.将CC与溶液转移至25 mL聚四氟乙烯内胆中,在200 ℃下水热反应24 h,反应结束后分别用去离子水和乙醇反复冲洗,并在60 ℃下干燥,即可得到CC@WS2NSs.
1.2.2 K2AZT 的合成 在H 型电解槽中,以CC@WS2NSs(0.5 cm×0.5 cm)作为阴极,CF(0.5 cm×0.5 cm)作为阳极,1.0 mol/L KOH与1.0 mol/L KOH+0.2 mol/L 5-AT分别作为阴极与阳极电解液,阴离子交换膜将两个反应室隔开,构建H 型双电极耦合体系.在10 mA/cm2下进行计时电位法测试(CP)15 h.反应结束后,加入乙酸乙酯,充分搅拌后,进行萃取旋蒸,得到黄色粉末,即为K2AZT.
1.2.3 结构表征 分别通过扫描电子显微镜、透射电子显微镜、X射线粉末衍射仪和X射线光电子能谱仪对CC@WS2NSs进行表征;以傅里叶变换红外光谱仪、差示扫描量热仪/同步热分析仪、核磁共振仪对合成K2AZT 粉末进行表征以得到红外光谱(IR)、热重/差示扫描量热(TG/DSC)曲线以及1H 和13C NMR 谱;K2AZT 溶液的Cu2+检测使用全谱直读等离子体发射光谱仪进行电感耦合等离子体发射光谱(ICP-OES)测试检验.
1.2.4 电化学测试 通过电化学工作站以三电极体系进行碱性HER催化性能测定,两电极体系构建耦合体系实现HER 与K2AZT 电合成的耦合.三电极体系以CC@WS2NSs、石墨棒、Hg/HgO(1.0 mol/L KOH)分别作为工作电极、对电极和参比电极,1.0 mol/L KOH作为电解液.两电极体系以CC@WS2NSs作为阴极、CF 作为阳极,分别以1.0 mol/L KOH 和1.0 mol/L KOH+0.2 mol/L 5-AT 作为电解液,构建H型双电极耦合体系.采用2.0 mV/s线性扫描伏安法(LSV)获得极化曲线,在100 kHz~0.1 Hz测试频率范围内获得电化学阻抗谱(EIS),采用循环伏安法(CV)测试在非法拉第电位区间不同扫描速率(v,2.5,5,10,25,50和100 mV/s)进行测量,并得到充电电流(ic),通过ic计算双电层电容Cdl(ic=vCdl),进而得到反应电化学活性表面积(ECSA)(ECSA=Cdl/Cs)[32](其中,Cs表示1 cm2真实表面积的平板标准电极的比电容),在碱性条件下的电容范围为Cs=0.022~0.130 mF/cm2[33],为了预测电化学面积,在碱性溶液中的Cs通常取0.040 mF/cm2[34].通过CP 测试评价催化剂的稳定性.所测电位由公式ERHE=EHg/HgO+0.059pH+0.098(其中,EHg/HgO表示以Hg/HgO作为参比电极三电极体系所测得的实际槽压)校正为可逆氢电极(RHE).通过计算Tafel斜率(η=a+blgj)[其中,η(V)为过电压;j(mA/cm2)为电流密度;a为常数参数;b为Tafel斜率]对样品的HER动力学速率变化进行评价.
2 结果与讨论
2.1 样品的结构与形貌表征
为了探究样品表面的结构特征,采用XRD 对合成材料的晶体结构进行测定(图1).可见,所合成的CC@WS2NSs 在2θ=9.3°,32.0°和56.6°处的特征峰与1T-WS2的(002),(022)和(008)晶面相对应[35],在2θ=21.9°和43.6°处的宽峰与CC 基底的特征峰相对应[36].此外,与WS2标准卡片(JCPDS No.08-0237)相比,CC@WS2NSs 相应的衍射特征峰出现了明显的左移,表明所制备的WS2为1T 相[35].与具有半导体性质的2H 相相比,具备金属性质的1T 相的WS2具有更优异的导电性和更丰富的活性位点,因而具有更优异的电化学性能[37,38].
样品CC@WS2NSs的表面微观结构特征采用SEM进行表征(图2).由图2(A)可见,WS2NSs均匀生长在二维CC表面;由图2(B)可见,WS2薄膜由交错堆叠的纳米片组成,WS2NSs均匀排列并且相互连接,构成粗糙的表面结构,增加了材料与电解质的接触面积,暴露了大量的活性位点[37].此外,CC提供自支撑结构,使得制备的CC@WS2NSs具有一定的机械稳定性,与粉末催化剂相比,具有不需黏结剂的优势,从而避免了因黏结剂引起的覆盖活性位点、增加界面阻力和降低传质能力的问题,此外,CC基底作为一种优良的导电基底,不仅能够提高电子导电率,还能暴露更多的活性位点以及抑制催化剂的团聚,使催化剂具备质量与电荷传递更快速的优点,从而具备更好的电催化性能[39,40].

Fig.1 XRD pattern of CC@WS2 NSs

Fig.2 Low(A) and high(B) resolution SEM images of CC@WS2 NSs
为了进一步确定CC@ WS2NSs 的表面结构特征,对样品进行了TEM 表征(图3).由图3(A)和(B)可见,WS2NSs 交错均匀生长在CC 基底上,由图3(C)的高分辨率TEM 照片可见,WS2的晶面间距为0.27 nm,与WS2的(100)晶面一致[41],进一步表明所合成为WS2.由高角度环形暗场-扫描透射电子显微镜(HAADF-STEM)照片[图3(D)]与相应的元素分布图[图3(E)和(F)]可见,W和S相互重叠与均匀分布,进一步表明了CC@WS2NSs的成功合成.

Fig.3 TEM(A,B),HRTEM(C),HAADF-STEM(D) images and elemental mapping of W(E),S(F) of CC@WS2 NSs
采用XPS对CC@ WS2的化学组成和表面电子结构状态进行分析.CC@WS2NSs XPS全谱图显示了W4f,S2p,C1s和O1s的特征峰[图4(A)],表明所制材料具有W,S,C和O元素,其中,C和O主要来自于空气以及CC 基底[41].如图4(B)W4f高分辨率XPS 谱图所示,在31.7 和33.8 eV 处的两个主峰分别与1T-WS2的对应,在32.1 和35.3 eV 处的两个主峰分别与2H-WS2的对应.在37.6 eV处的主峰与W氧化物的形成有关[42].此外,1T相WS2与2H相WS2相比整体向更低的结合能偏移,符合文献[43]报道的关于具备金属性质的1T-MX2材料的XPS研究结果,从而进一步表明所合成的WS2为1T相.在图4(C)中,161.2和162.3 eV 处的主峰分别与1T-WS2的对应[44].以上XPS谱图分析结果进一步表明所合成WS2为1T相,与XRD分析结果一致.
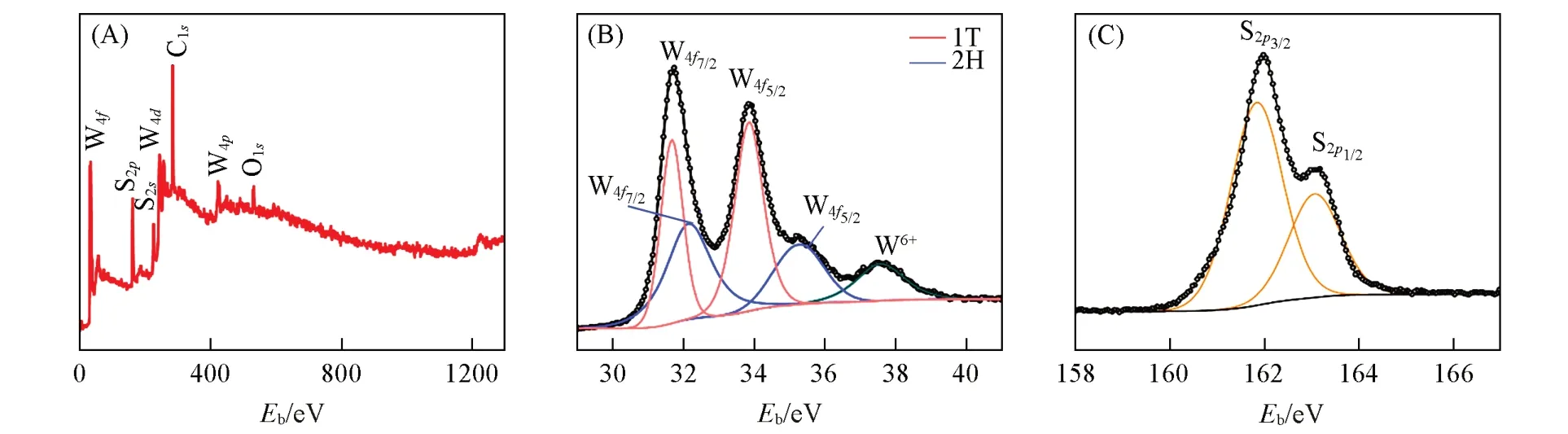
Fig.4 Survey(A),W4f(B) and S2p(C) XPS spectra of CC@WS2 NSs
2.2 碱性HER电化学测试
在1.0 mol/L KOH 溶液中使用LSV 极化曲线测试以评价CC@WS2NSs 的碱性HER 催化活性.如图5(A)所示,在-10和-100 mA/cm2的电流密度(j)下对应的过电位分别为280.1和395.7 mV.与其它过渡金属硫化物[如CoS2(327 mV),FeS2(300 mV),NiS2(301 mV)]在碱性环境-10 mA/cm2的电流密度下相比过电位更小[45~47],电化学反应速率更快、催化活性更好[48].
计算样品LSV 曲线中的Tafel 斜率,进而对样品的HER 动力学速率变化进行评价.通过LSV 曲线与Tafel方程(η=a+blgj,其中,n为过电势,j为电流密度,a和b为常数)计算CC@WS2NSs的Tafel斜率,碱性条件下HER通常被认为主要包含以下基元反应:
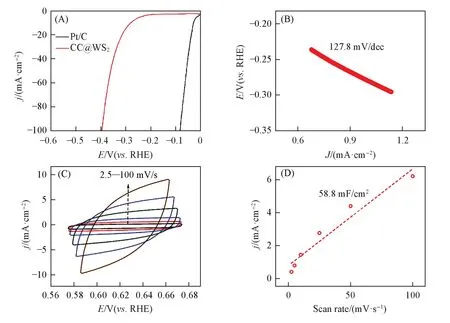
Fig.5 LSV(A) and Tafel(B) curves of CC@WS2 NSs,CV curves of CC@WS2 NSs with various scan rates(2.5,5,10,25,50,100 mV/s)(C),Cdl date for the CC@WS2 NSs(D)
通过计算Tafel斜率可以判断速率决定步骤,通常认为Tafel斜率分别为120,40与30 mV/dec时反应的速率决定步骤分别为Volmer,Heyrovsky 与Tafel[49].如图5(B)所示,CC@ WS2NSs 的Tafel 斜率为127.8 mV/dec,因此其碱性HER 过程中Volmer 步骤是速率决定步骤,表明缓慢的水吸附/解离过程(Volmer)控制了反应的动力学速率.
ECSA 是评估HER 动力学性能的另一项指标.采用CV 在不同扫描速率(2.5,5,10,25,50 和100 mV/s)下的非法拉第电位范围[图5(C)],测量Cdl以计算ECSA,进一步评价HER 催化性能.如图5(D)所示,CC@WS2NSs 的Cdl为58.8 mF/cm2,带入计算得ECSA=1470 cm2,表明CC@WS2NSs 有较大的ECSA,能够暴露更多的活性位点,促进碱性HER过程的动力学反应,提高材料的催化活性.
稳定性是电催化剂的另一个重要评价标准.采用多次CV测试后,对样品再次进行LSV测试,比较循环前后LSV 曲线重合程度和CP 曲线,以及对CP 后催化剂进行XRD,XPS 和SEM 表征,评价CC@WS2NSs 的长期催化的能力.循环2000 次前后LSV 曲线重合度较高,表明CC@WS2NSs 碱性条件下具有较高的稳定性[图S1(A),本文支持信息].在-0.576~0.324 V(vs.RHE)电位范围,-10 mA/cm2的电流密度下,过电位在超过50 h持续反应后没有出现明显的变化[图S1(B)],表明CC@WS2NSs具有良好的长期催化能力.CP 前后的XRD 谱图无明显变化[图S1(C)],表明WS2仍保持1T相结构未发生变化.为了进一步研究CP之后材料化学成分的变化,对CP后的材料进行XPS测试[图S1(D)],反应后元素组成无变化,进一步证实CC@WS2NSs具有良好的稳定性.CP后采用SEM表征以探究结构稳定性[图S1(E)和(F)],CP 50 h 后WS2NSs 仍均匀生长在碳布上,并且保持原有的3D 片状结构,表明了CC@WS2NSs 具有良好的结构稳定性.以上结果均表明了CC@WS2NSs 在碱性条件下经历长时间反应后仍具有良好的碱性HER催化稳定性与结构稳定性.
2.3 K2AZT电合成协同作用机理
Mishra等[50]和Kabanda等[51]证明了羟基自由基(·OH)可以从氨基中捕获单个氢原子与硫脲和硒脲反应的氢原子转移机理,随后·OH 在5-AT 光催化合成5,5′-偶氮四唑盐的激活中的重要作用也被证实[24],鉴于此,以添加·OH自由基清除剂的方式研究了·OH在介导K2AZT电合成中的激活作用.
以CF,Pt,Hg/HgO分别作为工作电极、对电极和参比电极,1.0 mol/L KOH+0.2 mol/L 5-AT+0.1 g 2,2,6,6-四甲基哌啶氧化物(TEMPO)或10 mL异丙醇作为电解液构成三电极体系分别进行LSV测试,探究K2AZT电化学合成机理.分别添加·OH自由基清除剂TEMPO与异丙醇,其LSV曲线如图6(A)和(B)所示,添加·OH自由基清除剂后,K2AZT电合成活性出现明显的衰减.表明·OH自由基清除剂对于K2AZT的电合成有抑制作用,即·OH参与了K2AZT的电合成反应过程.而在有机合成过程中,化合物的转化通常是由反应中间体(如自由基)所驱动,因此·OH 对于5-AT 氧化偶联形成K2AZT 具有激活作用[25],而·OH 常常在水氧化过程中产生[52].鉴于以上分析,K2AZT 的合成作用机理如图6(C)和(D)所示,5-AT在碱性环境中通过电化学的作用发生脱氢形成5-AT-,随后5-AT-与水氧化过程中产生的·OH结合发生反应并形成具有活性位点的*5-AT-,随后具有活性位点的相邻两个*5-AT-发生氧化偶联反应形成K2AZT.从而实现5-AT 的氧化偶联与水氧化的协同作用,验证了K2AZT 的电合成协同作用机理[27].
2.4 全解水与K2AZT电合成的耦合
所制备的CC@WS2NSs具有优异的碱性HER催化稳定性,然而动力学缓慢的OER反应阻碍了全解水过程中氢的高效生产,因此用具有热力学较低氧化电位的5-AT氧化偶联反应取代动力学缓慢的四电子反应OER,从而建立一种新的耦合体系.

Fig.7 LSV curves of different electrodes in 0.2 mol/L 5-AT concentration(A),LSV curves of CF electrode in different 5-AT concentration(B),schematic diagram of the H-type two-electrode coupling system using CC@WS2 NSs as the cathode and CF as the anode(C),LSV curves of electrosynthesis of K2AZT and overall water splitting in the coupling system(D),CP curve of the coupling system at -10 mA/cm2(E),optical photos of the coupling system before and after reaction(F),cell voltage compared to previously reported water splitting electrocatalysts at a current density of 10 mA/cm2(G)
以CF、泡沫镍(Ni)和钛片(Ti)分别作为工作电极,Pt 丝作为对电极,Hg/HgO 作为参比电极,1.0 mol/L KOH与不同浓度5-AT作为电解液构成三电极体系,分别进行LSV测试,探究K2AZT电化学合成的最佳电极和最佳浓度.如图7(A)所示,在10 mA/cm2的电流密度下分别以CF,Ti,Ni作为工作电极的三电极体系槽压分别为1.32,1.47和1.53 V,CF表现出最低槽压,即CF表现出最佳的电催化性能,CF对K2AZT电化学合成具有较高的活性.因此,可选用CF作为耦合体系的阳极材料.以CF作为三电极体系的工作电极,在不同5-AT 浓度(0,0.1,0.2,0.4,0.6 mol/L)进行LSV 测试,探究5-AT 浓度对于K2AZT电化学合成的影响.如图7(B)所示,随着5-AT浓度的增大,相同电流密度下槽压先降低后升高,这是由于随着浓度的升高,合成物的质量迁移速率逐渐大于反应物的质量迁移速率,从而在电极表面逐渐形成有机层,阻碍了5-AT分子接近电极表面发生反应,进而使得槽压升高[53].在电流密度为10 mA/cm2时,浓度为0.2 mol/L的5-AT表现出1.29 V的最低槽压,即在三电极体系下,CF电极与0.2 mol/L 5-AT能够使体系达到最低槽压,因此,可选用CF作为最佳电极、0.2 mol/L作为5-AT的最佳溶液浓度进行后续测试.
以CC@WS2NSs 作为阴极电极,CF 作为阳极电极,1.0 mol/L KOH 与1.0 mol/L KOH+0.2 mol/L 5-AT 分别作为阴极与阳极电解液,以阴离子交换膜将两个反应室隔开,构建H 型双电极耦合体系[图7(C)].以CF为阳极电极,在1.0 mol/L KOH+0.2 mol/L 5-AT阳极电解液与不添加5-AT条件下进行LSV 测试,如图7(D)所示,在电流密度为10 mA/cm2时,0.2 mol/L 5-AT 耦合体系表现出1.65 V 的槽压,比未添加5-AT 的CC@WS2NSs/CF 全解水体系(1.87 V)低220 mV,与近年报道的全解水制氢体系相比具有更低的槽压[图7(G)和表S1,见本文支持信息],充分证实了5-AT氧化偶联反应取代OER以实现更低槽压电解水制氢的可行性.此外,相比于未添加5-AT 的CC@WS2NSs/CF 全解水体系,0.2 mol/L 5-AT耦合体系更接近于Pt/IrOx体系在电流密度为10 mA/cm2时的槽压(约1.5 V)[54].
如图7(E)所示,在电流密度为-10 mA/cm2下,该耦合系统在超过15 h的持续通电反应中,过电势没有产生明显的变化,表明了该耦合体系具有优异的稳定性.在反应过后,阳极侧溶液由无色变为黄色,且CF电极表面没有气泡[图7(F)],为了确定溶液中是否存在Cu2+,对溶液进行ICP-OES测试,未检测出Cu2+,证明无CuAZT的合成.以上现象初步表明K2AZT的成功合成且OER未发生,即5-AT氧化偶联反应成功取代了OER反应.同时,在阴极侧CC@WS2NSs表面出现气泡[图7(F)],表明H2在阴极侧生成.
对溶液浓缩后得到的K2AZT 进行FTIR 表征[图8(A)],可见,使用传统有机合成方法合成的标准K2AZT在1395和734 cm-1附近具有最强的吸收峰,并分别对应于偶氮四唑环中的C—N3和C—N2不对称拉伸模式[55].3368和3277 cm-1处的特征峰归属为结晶水的O—H键的伸缩振动;1665 cm-1处的峰归属为偶氮四唑环的N=N 键和N—N 键的伸缩振动;1197,1155 和1055 cm-1处的峰归属为偶氮四唑环的C—N键的伸缩振动;775和734 cm-1处的峰归属为四唑环的振动[56,57],通过电合成方式合成的K2AZT和标准K2AZT所表现的最强特征吸收峰一致,表明K2AZT的成功合成.为了进一步证明所合成的化合物为K2AZT,对合成的产物进行1H NMR和13C NMR测试,如图8(B)所示,δ=4.67处出现特征峰归因于在核磁过程中需要使用氘代水(D2O)溶剂,K2AZT 吸附了水.如图8(C)所示,δ=172.06 处出现特征峰,对应于K2AZT的四唑碳.以上结果进一步表明在阳极5-AT氮氮氧化偶联合成了K2AZT.除此之外,对K2AZT 进行热分析研究[图8(D)和(E)].由图8(D)可见,当温度达到188.54 ℃时,K2AZT 开始失重,并在234.14 ℃达到首个峰值,并且少量失重,表明K2AZT发生了部分升华.在286.82 ℃达到最高峰,失重最明显,说明K2AZT 发生了热分解[56,58].由图8(E)可见,在241.47 ℃出现吸热峰,该温度与图8(D)所分析的吸热峰温度相近,因而可能是K2AZT的升华所致,在275.54 ℃出现了放热峰,证明发生了K2AZT的热分解.综上,该耦合系统在阳极通过绿色电化学的方式合成了K2AZT,避免了传统合成方式的安全与成本问题,同时,在阴极具有催化H2生成的能力.
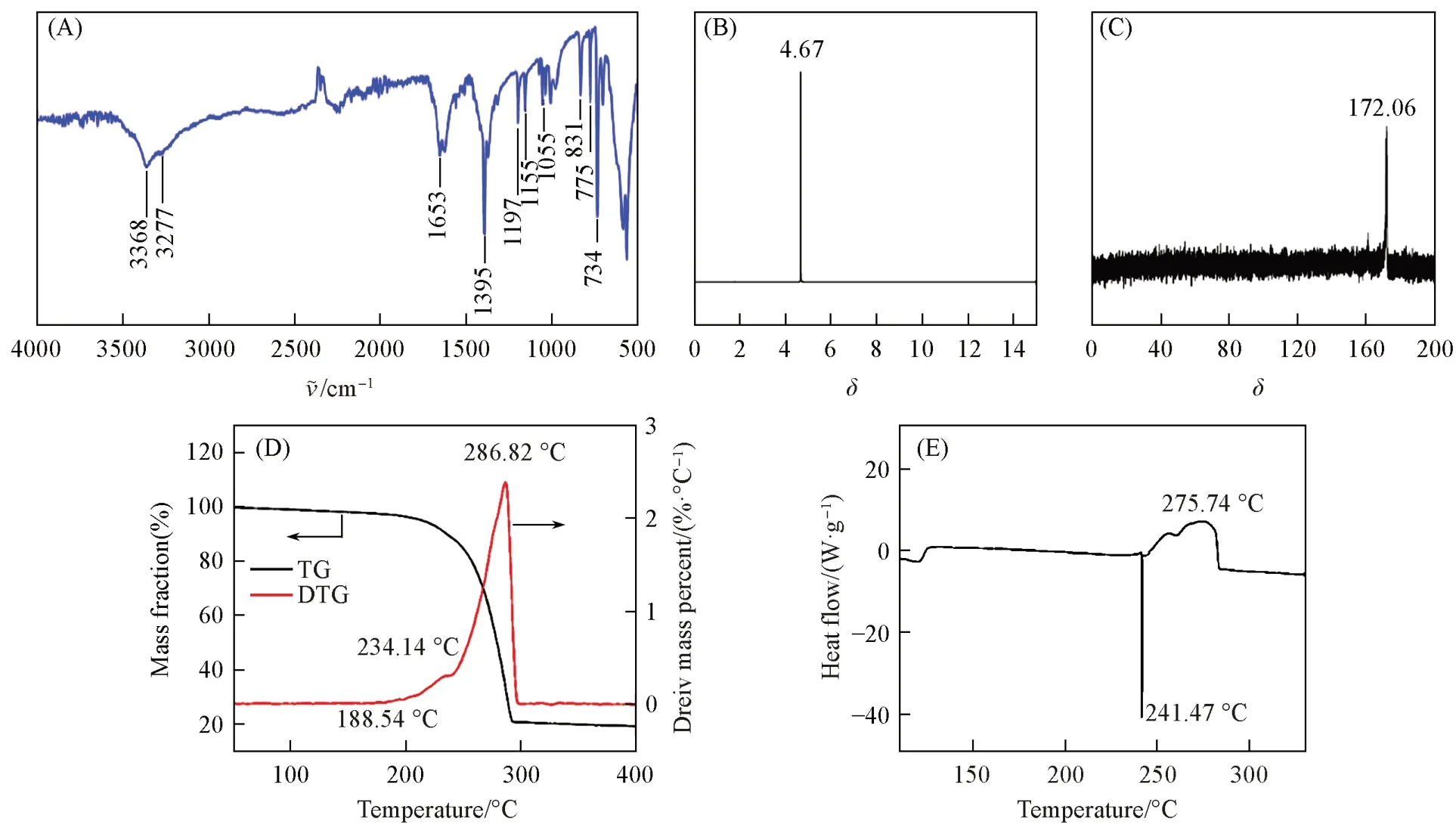
Fig.8 FTIR diagram of electrosynthesized K2AZT(A),1H NMR(B) and 13C NMR(C) spectra of the coupling products(K2ATZ),TG-DTG(D) and DSC(E) curves of K2AZT at a heating rate of 10 ℃/min
3 结论
建立了一种新的耦合体系,在阳极以CC@WS2NSs作为碱性HER催化剂,同时以5-AT氧化耦合反应取代OER 反应,在降低碱性电解水槽压制得氢气的同时,通过绿色电化学的方式制得了含能材料K2AZT.CC@WS2NSs阴极催化剂通过一步水热法制得,在超过50 h持续反应后没有出现明显的变化,表现出一定的催化性能与优异的稳定性.以添加·OH自由基清除剂的方式,验证了5-AT氧化偶联与水氧化反应协同作用机理.耦合体系在电流密度为10 mA/cm2下,表现出1.65 V 的槽压,相比于CC@WS2NSs/CF全解水体系(1.87 V)降低了220 mV,经历15 h持续反应后仍表现出良好的催化活性,表明该体系优异的催化性能与稳定性.同时,在阳极侧通过电化学合成的方式制得K2AZT,从而以更环保、安全的方式实现了K2AZT的绿色合成.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20230347.
猜你喜欢
心肺血管病杂志(2019年1期)2019-04-22
电镀与环保(2017年5期)2017-12-19
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年2期)2017-01-20
浙江农业科学(2016年11期)2016-05-04
当代化工研究(2016年9期)2016-03-20
广州大学学报(自然科学版)(2015年4期)2015-12-23
中国塑料(2015年8期)2015-10-14
