
关节炎中滑膜增生的研究进展
2023-12-21 09:57刘海港郭州张雄孙凯郭风劲
骨科 2023年6期
刘海港 郭州 张雄 孙凯 郭风劲
关节炎是以关节和邻近结构的炎症为主要特征的一组多样化疾病,可能导致疼痛、畸形和残疾,成人和儿童均可受累。关节炎包括类风湿性关节炎、骨关节炎、化脓性关节炎(感染性)、创伤后关节炎和血友病性关节炎等重要类型,其中类风湿性关节炎和骨关节炎最为常见。
滑膜是覆盖在关节除软骨以外的内表面的结缔组织薄膜,由表面的衬里层和深部的衬里下层构成[1-3]。滑膜衬里层在正常状态下排列着1~3层细胞,分别是巨噬细胞样滑膜细胞,也称作A型滑膜细胞,以及成纤维细胞样滑膜细胞(fibroblast-like synoviocyte,FLS),即B型滑膜细胞[4-6]。在衬里下层的深层滑膜分布着大量的脂肪细胞、肥大细胞、成纤维细胞、蛋白聚糖和弹力纤维。在关节炎症状态下,滑膜衬里层细胞层数增多、炎症细胞浸润增加、滑膜新生血管形成和纤维化、滑膜充血肿胀,最终引起滑膜的厚度明显增加,这一病理现象称为滑膜增生[7-8]。滑膜增生是类风湿性关节炎、血友病性关节炎和骨关节炎等关节炎性疾病所共有的重要病理改变,在这些疾病的进程中起到了关键的作用。
一、文献检索策略选择
本文以“滑膜增生”、“关节炎”、“滑膜增殖”为关键词在中文数据库中国知网、万方数据库中展开检索;以“synovial hyperplasia”、“arthritis”为关键词在国际期刊数据库PubMed、Web of Science 中进行检索。文献纳入标准:①纳入文献类型为发表在学术期刊上的论著和综述;②研究内容相似的文献中,优先选择具有更高研究证据级别的文献;③研究内容同时涉及关节炎和滑膜增生。排除标准:①除中文、英文之外的其他语种的文献;②文献类型为评论和会议论文;③重复发表的文献和无法获取全文的文献。本文一共检索到中文文献212篇(中国知网153篇,万方数据库59篇),英文文献974篇(PubMed 384篇,Web of Science 590篇)。经筛选后最终只有45篇被纳入,其中中文7篇、英文38篇(图1)。
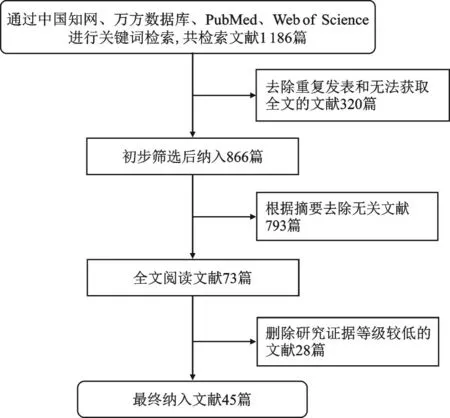
图1 文献纳入流程图
二、关节炎中滑膜增生的机制
探究滑膜增生发生机制最重要的问题是明确滑膜增生的细胞来源。目前,学界认为滑膜增生主要与成纤维样滑膜细胞增殖、炎症与免疫细胞浸润和来自于骨髓迁移的间充质干细胞有关。滑膜增生并不是任何单独细胞种类的作用,而是滑膜中几乎所有细胞群之间复杂的相互作用的结果。
(一)FLS增殖与凋亡失衡
在类风湿性关节炎和骨关节炎中,滑膜增生是滑膜衬里层FLS 过度增殖和凋亡减少造成的。研究者通过对类风湿关节炎、骨关节炎病人的滑膜组织免疫组织化学染色,发现滑膜衬里层FLS的标志物Ⅰ型前胶原的表达较高,而巨噬细胞的标志物集落分化抗原68(cluster of differentiation 68,CD68)表达很少[9]。同时细胞增殖的标志物细胞增殖核抗原(proliferating cell nuclear antigen,PCNA)和原癌基因c-Myc也在滑膜细胞衬里层出现了明显的高表达。进一步研究发现,表达PCNA和c-Myc的细胞对CD68呈阴性表达,这一现象说明了在滑膜衬里层发生增殖的细胞主要是FLS,而非血液来源的巨噬细胞。此外,从滑膜周围血管中迁移到滑膜的单核细胞,在经历成熟过程转变为巨噬细胞后,其增殖能力会出现明显的降低。有研究发现,在滑膜组织培养中,那些存活三代以上的滑膜细胞中只有不到1%为巨噬细胞[10],因此排除了巨噬细胞样滑膜细胞通过分裂增殖引起滑膜增生的可能。
FLS 的凋亡减少也被认为有助于滑膜组织的过度增生。转录激活因子3(signal transduction and activator of transcription 3,STAT3)是细胞内一种重要的信号转导蛋白和转录因子。在炎症状态下,激活的STAT3能通过上调B细胞淋巴瘤-XL(B-cell lymphoma-extra large,Bcl-xL)或下调B 淋巴细胞瘤-2 基因(Bcl-2)关联X 蛋白(Bcl-2-associated X protein,Bax)来抑制caspase-3 活性从而起到抗凋亡作用[11]。Shen等[12]研究表明,持续活化的STAT3保护成纤维细胞免受血清饥饿诱导的凋亡,而特异性失活STAT3则会诱导滑膜细胞凋亡[13]。肿瘤蛋白p53基因(tumor protein p53,TP53)在细胞凋亡调控网络中扮演着关键角色,它能够靶向影响内源性和外源性凋亡途径的关键基因,包括细胞色素c、Bax、Fas 和caspase-8等[14]。有研究表明,滑膜细胞中p53突变可导致类风湿性关节炎滑膜细胞凋亡抵抗和病理性细胞过度存活[15]。为了进一步证明在类风湿性关节炎的FLS 中,突变的p53是否具有抗凋亡的特性,有研究人员设计了一个表达p53源融合肽的腺病毒载体,并在胶原诱导性关节炎大鼠关节中注射该载体。他们发现注射腺病毒的关节中关节指数和组织学评分降低,滑膜细胞密度减少,凋亡细胞数量增加,并且p53细胞凋亡调节因子(p53 upregulated modulator of apoptosis,PUMA)表达水平更高[16]。另外,他们还发现在类风湿性关节炎滑液成纤维细胞中,PUMA的表达水平较低。这一观察为p53 介导的细胞凋亡途径受损提供了另一种解释[17]。
(二)血管来源的炎症细胞浸润
另外一种观点认为增生的滑膜与周围血管的炎症细胞浸润有关。Lalor等[18]通过对类风湿性关节炎和骨关节炎病人滑膜标本进行了Ki-67 免疫染色以确定增殖细胞的来源。Ki-67 是一种细胞核内蛋白质,通常用作细胞增殖的生物标志物,其染色的阳性结果通常表明这些细胞正处于活跃的细胞分裂状态。研究者发现在类风湿性关节炎和骨关节炎滑膜衬里层中Ki67阳性染色的细胞占总细胞数的百分比小于0.03%,而在炎症细胞浸润区域中却有大量的Ki67阳性细胞存在。这说明增生的滑膜衬里层并没有通过局部细胞分裂产生,而是由炎症细胞浸润而来。Yudoh 等[19]研究发现,类风湿性滑膜炎滑膜淋巴细胞中的端粒酶活性水平与PCNA表达、滑膜内层增生的程度和微血管增生、血管周围浸润高度相关。具有高端粒酶活性的滑膜浸润淋巴细胞可能产生多种细胞因子、生长因子,如白细胞介素-1(interleukin-1,IL-1)、白细胞介素-6(interleukin-6,IL-6)或肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α),刺激滑膜细胞增生。有趣的是,Neumann等[20]报道滑膜增生组织内新血管生成增加也能促进炎症细胞涌入滑膜。
(三)骨髓来源的间充质细胞迁移
在关节炎中,骨髓来源的间充质细胞的流入很可能有助于滑膜血管翳的形成[21]。研究人员发现在胶原诱导的类风湿性关节炎的动物模型中,大量骨髓来源的间充质细胞迁移到滑膜,占据了成纤维样滑膜细胞的30%以上,这在健康滑膜组织中几乎检测不到[21]。这些间充质细胞中核因子κB(nuclear factor kappa-B,NF-κB)通路特异性激活,可抑制其多向分化并促进增殖[21]。在关节面受损引起的创伤性骨关节炎模型中,研究者也观察到了类似的骨髓源性细胞的浸润导致滑膜增生的现象,并推测骨髓间充质干细胞通过骨管进入到滑膜和关节间隙中[22]。至于骨髓间充质细胞募集到滑膜中的深层机制,目前有文献报道可能与关节中胎盘生长因子的水平升高有关[23]。此外,Galligan 等[24]报道循环成纤维细胞在类风湿性关节炎中被激活,高表达各种趋化因子的配体,由此被关节炎滑膜中的大量趋化因子招募并迁移到滑膜中,有助于滑膜的增生。
三、关节炎中滑膜增生的影响
滑膜的功能包括润滑关节面,减少运动时的摩擦以及营养和免疫功能。在健康的关节中,FLS 产生关节润滑剂,例如透明质酸和润滑素。FLS还通过产生各种基质成分(例如Ⅳ型胶原蛋白)来帮助塑造细胞外基质。在类风湿性关节炎中,关节的滑膜衬里层大幅扩张并转变为侵袭性增生性血管翳。此前的研究表明,异常增生的FLS表现出与肿瘤细胞类似的侵袭性特征[25]。Laragione 等[26]的研究显示哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号通路参与FLS 侵袭的调节,mTOR 的抑制剂雷帕霉素显著减少FLS 的侵袭。FLS 在与浸润到关节的免疫细胞接触时,或在暴露于生长因子和炎症细胞因子(例如TNF-α)时,会产生许多趋化因子、促炎因子、基质金属蛋白酶和组织蛋白酶。其中基质金属蛋白酶是降解细胞外基质和软骨的重要因素。促炎因子(例如IL-6)、趋化因子(例如CXC-趋化因子配体10)和促血管生成因子会进一步加重和维持关节炎症[27]。正如上文中提到的,滑膜血管增生也会导致炎症细胞的浸润增多而加速炎症反应[28]。
骨关节炎与类风湿性关节炎中的FLS 还通过促进破骨细胞生成和抑制骨修复而导致骨侵蚀[29]。破骨细胞形成的关键介质是NF-κB 受体活化因子配体(receptor activator of nuclear factor-kappa B ligand,RANKL),它通过与NF-κB受体活化因子(receptor activator of nuclear factor-kB,RANK)的结合,直接诱导破骨细胞前体的分化成熟,并促使成熟的破骨细胞吸收骨组织。RANKL 的功能受到破骨细胞抑制因子(osteoclastogenesis inhibitory factor,OPG)的竞争性调控[30]。为了探讨FLS 中RANKL 表达对破骨细胞生成的直接作用,研究者使用FLS特异性RANKL敲除小鼠模型,发现胶原诱导的关节炎模型中破骨细胞生成和骨侵蚀明显减少。研究者还证明了在软骨细胞或T细胞中特异性敲除RANKL对炎症性关节炎期间破骨细胞生成、骨侵蚀和软骨破坏没有影响[30]。Croft等[31]发现在血清转移诱导的关节炎模型中,增生的FLS也表达出较高的RANKL 水平,同时羟基磷灰石基质的吸收显著增加。滑膜中募集的间充质干细胞又被称为滑膜衍生基质细胞。研究者通过比较健康受试者和骨关节炎病人滑膜衍生基质细胞诱导正常外周血单个核细胞(peripheral blood mononuclear cell,PBMC)分化为破骨细胞的能力,发现两种滑膜衍生基质细胞均刺激PBMC 形成破骨细胞。其中健康受试者的滑膜衍生基质细胞与PBMC 的Transwell 共培养产生的破骨细胞样细胞不活跃,而骨关节炎衍生的滑膜衍生基质细胞刺激PBMC 分化为活跃的破骨细胞并介导软骨的损伤[32]。
四、滑膜增生的信号调控通路
(一)类风湿性关节炎滑膜增生的信号调控通路
RANKL/NF-κB通路被认为在类风湿性关节炎滑膜细胞增殖中发挥重要作用[33]。Zhou等[34]分别通过MTT和TUNEL实验验证了RANKL抑制剂对于体外培养FLS的抑制增殖和促凋亡作用。为了进一步研究RANKL抑制剂影响FLS存活的机制,研究者对NF-κB和caspase-3的mRNA和蛋白水平进行了检测。检测结果显示,RANKL 抑制剂明显降低了它们的表达水平。这说明RANKL抑制剂可能通过抑制NF-κB信号通路来抑制FLS的增殖并促进凋亡(图2)。
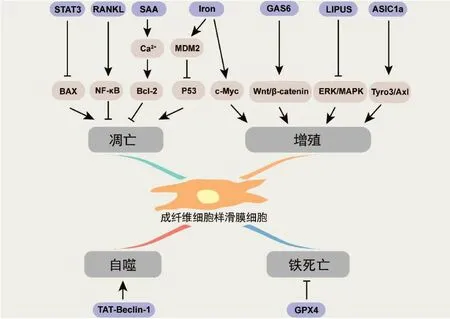
图2 成纤维细胞样滑膜细胞增殖、凋亡、自噬与铁死亡的调控通路示意图 在关节炎中,各种分子或理化因素通过相应信号通路调节成纤维细胞样滑膜细胞的增殖、凋亡、自噬与铁死亡等细胞事件(绘图作者:刘海港)
酸敏感离子通道1a(acid-sensing ion channel 1a,ASIC1a)被报道通过下游的丝裂原活化蛋白激酶(Mitogen-activated protein kinase,MAPK)/细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)通路刺激FLS 增殖[35]。研究人员发现细胞外酸化增强了FLS 的集落形成能力,并增加了PCNA 和Ki67 的表达。而使用ASIC1a 的特异性抑制剂可以逆转这种效应,表明细胞外酸化通过激活ASIC1a促进了FLS的增殖。同时被ASIC1a的特异性抑制剂下调的还有加速纤维肉瘤(rapidly accelerated fibrosarcoma,RAF)蛋白和ERK 信号。添加ERK抑制剂(U0126)后进一步抑制了FLS的增殖,这说明ERK/MAPK 信号通路有助于ASIC1a 激活促进的FLS增殖过程[36](图2)。
生长停滞特异性蛋白6(growth arrest-specific 6,GAS6)被发现在RA病人的滑膜中表达上调。有研究表明GAS6通过与受体酪氨酸激酶(Tyrosine-protein kinase receptor,Tyro3/Axl)相互作用,诱导人FLS 增殖[37]。受体酪氨酸激酶Tyro3的缺失可抑制关节炎中的滑膜增生和破骨细胞分化(图2)。
血清淀粉样蛋白A(serum amyloid A,SAA)是一种主要的急性期反应物,已被证明可以介导促炎细胞反应。在一项研究中,研究者发现SAA 促进人FLS 的增殖[38]。此外,SAA还可以保护类风湿性关节炎FLS 免受血清饥饿、抗Fas、IgM和硝普钠诱导的细胞凋亡。进一步探究发现,SAA对FLS增生的影响是由细胞内钙水平增加以及ERK 和蛋白激酶B(protein kinase B,PKB/Akt)激活引起的,从而导致细胞周期蛋白D1和Bcl-2表达升高(图2)。
(二)骨关节炎滑膜增生的信号调控通路
经典Wnt/β-连环蛋白(Wnt/β-catenin)与细胞增殖、纤维化、胚胎骨骼形成、组织修复和关节稳态密切相关[39]。先前的一项研究报道,骨关节炎内侧半月板不稳定模型(destabilization of the medial meniscus,DMM)小鼠和骨关节炎病人的FLS 中Wnt/β-catenin 信号被激活,Wnt/β-catenin 信号的药理抑制可减弱FLS 的增殖[40]。最近的研究[41]发现低强度脉冲超声(low-intensity pulsed ultrasound,LIPUS)通过Wnt/βcatenin 途径来调节FLS 增殖和纤维化反应。研究者在施加LIPUS 干预的情况下,用Wnt3a 重组蛋白刺激FLS 后检测Wnt/β-catenin 信号通路的表达变化。实验结果表明,LIPUS抑制FLS中Wnt/β-catenin信号的激活和纤维化标志物(α-平滑肌肌动蛋白、结缔组织生长因子和胶原蛋白Ⅰ)的表达。此外,Wnt3a重组蛋白刺激FLS后,细胞增殖水平和细胞活力明显增加,这恰好与LIPUS处理引起的效应相反(图2)。
D-异构体TAT-Beclin-1是一种有效的自噬诱导剂,可以通过激活自噬通路的重要分子Beclin-1而在体外具有更高的自噬激活潜力。研究者发现在模拟创伤性骨关节炎的小鼠DMM模型中,TAT-Beclin-1可以呈浓度依赖性地诱导严重的滑膜增生、纤维化并伴有自噬关键标志物表达增加和凋亡关键标志物表达减少[42]。同时,TAT-Beclin-1干预后滑膜内膜还出现了成纤维细胞活化以及炎症细胞浸润增加。这些现象提示D-异构体TAT-Beclin-1 诱导滑膜增生可能是通过促进FLS自噬和抑制其凋亡实现的(图2)。
(三)血友病性关节炎滑膜增生的信号调控通路
血友病性关节炎或血友病性关节病是血友病病人中一类常见的并发症,主要表现为反复的关节出血导致的滑膜增生、炎症以及软骨的进行性破坏。关节出血会引起FLS 增殖、肥大和新生血管生成。铁被认为在这一过程中发挥着重要的作用。在出血后,红细胞通常被滑膜巨噬细胞吞噬清除。由于滑膜巨噬细胞对血液的清除能力有限,大量红细胞分解产物含铁血黄素在其中沉积并进一步刺激了FLS 的增殖并诱导c-Myc 和小鼠双微体2(murine double minute 2,MDM2)癌基因的表达[43-44]。体外铁剂干预滑膜细胞也出现了类似的结果。MDM2 是一种癌蛋白,可以与p53 相互作用引起其失活,抑制p53 依赖性细胞凋亡通路。而c-Myc 的增加可进一步促进滑膜细胞增殖[44](图2)。
有研究发现,在HA中FLS铁死亡受到明显抑制,表现为谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)在含铁血黄素沉积周围强烈表达[45]。GPX4是铁死亡的重要标志物,能够催化脂质过氧化物的还原反应从而抑制铁死亡的进行(图2)。此外,研究者还发现铁过载引起FLS 产生并释放IL-6 和TNF-α增加,进一步加剧了滑膜细胞增殖,这可能与其下游的磷脂酰肌醇3 激酶(phosphoinositide 3-kinase,PI3K)-Akt 通路或核因子E2 相关因子2(nuclear factor-erythroid 2-related factor 2,Nrf2)通路被激活相关。这些结果提示了铁死亡诱导剂和TNF-α抑制剂对血友病性关节炎滑膜增生的潜在治疗价值。
五、总结与展望
滑膜增生是关节炎病人的常见病理表现,发生机制包括FLS 的不断增殖、凋亡失衡、炎症细胞的浸润以及来自骨髓的间充质细胞的迁移。这一过程严重影响了关节的正常功能,导致了关节润滑物和基质成分的紊乱,从而促进了炎症反应和骨质侵蚀。尽管类风湿性关节炎、骨关节炎和血友病性关节炎等关节疾病都表现出滑膜增生,但是对于滑膜增生的研究目前更多集中于类风湿性关节炎,这可能是因为滑膜增生是类风湿性关节炎的核心病理改变之一。作为滑膜衬里层重要的细胞成分之一,FLS的凋亡与增殖逐渐成为滑膜增生机制研究的焦点。我们在前文着重讨论了RANKL/NFκB、ERK/MAPK、Wnt/β-catenin、MDM2/p53、Tyro3/Axl 等通路在调控FLS的凋亡与增殖中的作用。除此以外,铁死亡和自噬对FLS 的作用也是不可忽略的。我们课题组的前期研究中已经发现软骨细胞铁稳态失衡加速了小鼠骨关节炎和血友病性关节炎中的软骨退变。然而血友病性关节炎由于反复关节出血引起的铁沉积主要发生在滑膜组织,因此对铁在血友病性关节炎滑膜病变中的作用进行深入研究可能具有更大的价值。特别是目前针对滑膜如何代谢铁和转运铁这一关键领域的认识依然较少,亟待更多的研究来填补这一空白。总的来说,关节炎中的滑膜增生是一个复杂且关键的病理过程,涉及到关节中各种细胞的参与及相互作用,需要更深入地理解其调控机制以开发新的预防或治疗策略。
猜你喜欢
中国骨质疏松杂志(2024年2期)2024-03-19
中国特种设备安全(2021年5期)2021-11-06
中国骨质疏松杂志(2021年9期)2021-10-08
安全、健康和环境(2020年1期)2020-03-25
中国临床医学(2019年3期)2019-01-04
石油化工建设(2018年6期)2018-04-22
中国民族医药杂志(2016年2期)2016-05-14
中国民族医药杂志(2016年8期)2016-05-09
中国民族医药杂志(2016年8期)2016-05-09
石油化工建设(2015年1期)2015-12-01
