
多元有机酸自组装硅碳复合材料用于高效储锂
2024-01-06 04:52贲淼刘少卿陶丽娟李群寅张燕青刘建文
湖北大学学报(自然科学版) 2024年1期
贲淼,刘少卿,陶丽娟,李群寅,张燕青,刘建文
(1.湖北大学化学化工学院,湖北 武汉 430062; 2.湖北万润新能源科技股份有限公司, 湖北 十堰 442500)
0 引言
当前,锂离子电池广泛用于便携式电子设备和移动电话,在电动汽车等应用领域也显示出巨大的应用潜力。然而,锂离子电池仍然缺乏所需的能量储存水平,无法完全满足电动汽车等应用的需求[1]。硅具有很高的理论容量(约4 200 mAh/g)、相对较低的放电电压(<0.5 V vs. Li+/Li)和地壳储量丰富等特点[2],被认为是最有前途的锂离子电池负极材料之一。然而,硅在重复充放电循环过程中经历了巨大的体积变化(>300%),这会导致硅粒子的剧烈粉碎,从而导致电池容量在循环过程中迅速衰减[3]。此外,原始硅的低本征电导率显著抑制了其充放电行为。为了克服这些障碍并稳定固态电解质膜(SEI),人们进行了许多的尝试。这其中包括导电碳涂层封装硅[4],纳米结构工程[5-6],表面改性[7-9]、复合材料设计[10-12]、粘结剂优化[13-16]和电解质调制[17]。碳涂层是提高导电性、缓冲体积膨胀和稳定SEI的最有效策略之一[18-23]。过硅碳复合的方式已被研究和公认为提高电池性能的有效方法。碳基体不仅提高了Si基阳极的电子导电性,而且还起到缓冲作用,以适应体积变化,抑制机械应力。通过使用不同碳基体,可以有效地提高Si阳极的导电性和结构稳定性。各种碳材料,如碳纳米管(CNTs)[24-25],石墨烯[26]和碳纤维[27-28],已被证实对硅基负极材料的性能改善有明显的效果。然而,其中大多数材料都属于昂贵,且制作制备复杂。设计一种多核卵黄结构被证明是一种有效的改进策略,以便减弱硅在嵌锂过程中的体积膨胀。虽然这些研究在硅基材料方面取得了可喜的成果,但在工业和商业应用方面仍面临严峻的挑战,因此,探索一种简单的、低毒的、自组装的硅负极以减缓硅的体积变化意义重大。
在本研究中,我们使用自组装的方式制备材料,通过分子间的键合作用力形成稳定的结构。具体为使用3-氨基丙基三乙氧基硅烷(APTES)对纳米硅粉进行修饰,将修饰好的硅粉与不同的有机羧酸进行酰胺化反应,在将纳米硅颗粒交联到一起的同时,过量的羧酸材料在形成的颗粒表面形成一层碳涂层,增加其导电性和结构的机械稳定性。制备的多核卵壳结构硅碳负极材料,这种结构使得该电极材料在电流密度500 mA/g下,经过600圈的长循环下具有1 073.4 mAh/g的比容量,在2 A/g的电流密度下,初始比容量为751.2 mAh/g,在300圈循环后比容量维持在573.0 mAh/g,该材料制备方式为硅碳复合负极材料在低成本、多结构合成提供了一种新的思路。
1 实验部分
1.1 样品制备使用试剂有纳米硅粉(科路得,100 nm),APTES(3-氨基丙基三乙氧基硅烷),柠檬酸(缩写为nms),甘氨酸(缩写为gas),反丁烯二酸(缩写为fds)。合成方法图1所示。取9个50 mL的干净烧杯,分别加入0.2 g的纳米硅粉和30 mL的乙醇,超声分散30 min,之后分别加入1 mL的APTES,搅拌8 h。随后按照硅碳元素质量比10%、20%、30%的比例加入3种酸材料,一共得到9种材料,分别记录在表1中。将上述材料继续搅拌8 h,分散均匀后,转移到烘箱中,80 ℃下6 h干燥,将干燥后材料在氩气保护下以5 ℃/min的升温速率升温到800 ℃,保持3 h后自然退火,最终得到9种复合材料,也记录于表1中,进行电池装配及性能测试。

表1 本研究各步材料记录表
1.2 电池装配将上述9种复合材料分别与乙炔黑、羧甲基纤维素钠按照质量比8∶1∶1在去离子水中分散为浆液,之后将浆料涂布在铜箔上,在80 ℃下干燥12 h,得到硅碳负极材料的极片,将极片切成直径8毫米的极片。所使用的电解液成分为商用电解液,采用六氟磷酸锂(LiPF6)为电解质盐,在碳酸乙烯酯(EC)、碳酸二甲酯(DMC)和碳酸二乙酯(DEC)的体积比为1∶1∶1的混合溶剂分散,浓度为1 mol/L。使用锂金属为对电极,制备成半电池。电池性能通过蓝电电池测试系统在0.01 V到3 V进行电池性能测试,循环伏安法(CV)和电化学阻抗在电化学工作站(CHI 660E)上进行。
1.3 测试条件红外测试:使用红外光谱测试仪在400~4 000 cm-1的扫描范围下进行测试。
XRD(X射线衍射测试)测试:使用X射线衍射测试仪在2θ范围为5°~80°的范围下测试。
热重测试:以10 ℃/min的升温速度在空气氛围下进行测试,测试的稳定范围为30~800 ℃。
拉曼测试:使用532 nm的激光发射器在0~2 500 cm-1的扫描范围下测试。
粒度测试:使用激光粒度仪对材料进行粒度测试。
2 结果与讨论
2.1 合成反应通过自组装的方式,将修饰好的纳米硅粉和多元的有机羧酸组合起来,是一种有效减弱硅膨胀效应的硅碳复合材料制备方式,合成反应方程式如图2所示。甘氨酸、反丁烯二酸、柠檬酸,由于自身所带的羧酸基团数量的不同,分别形成如下不同的复合材料。
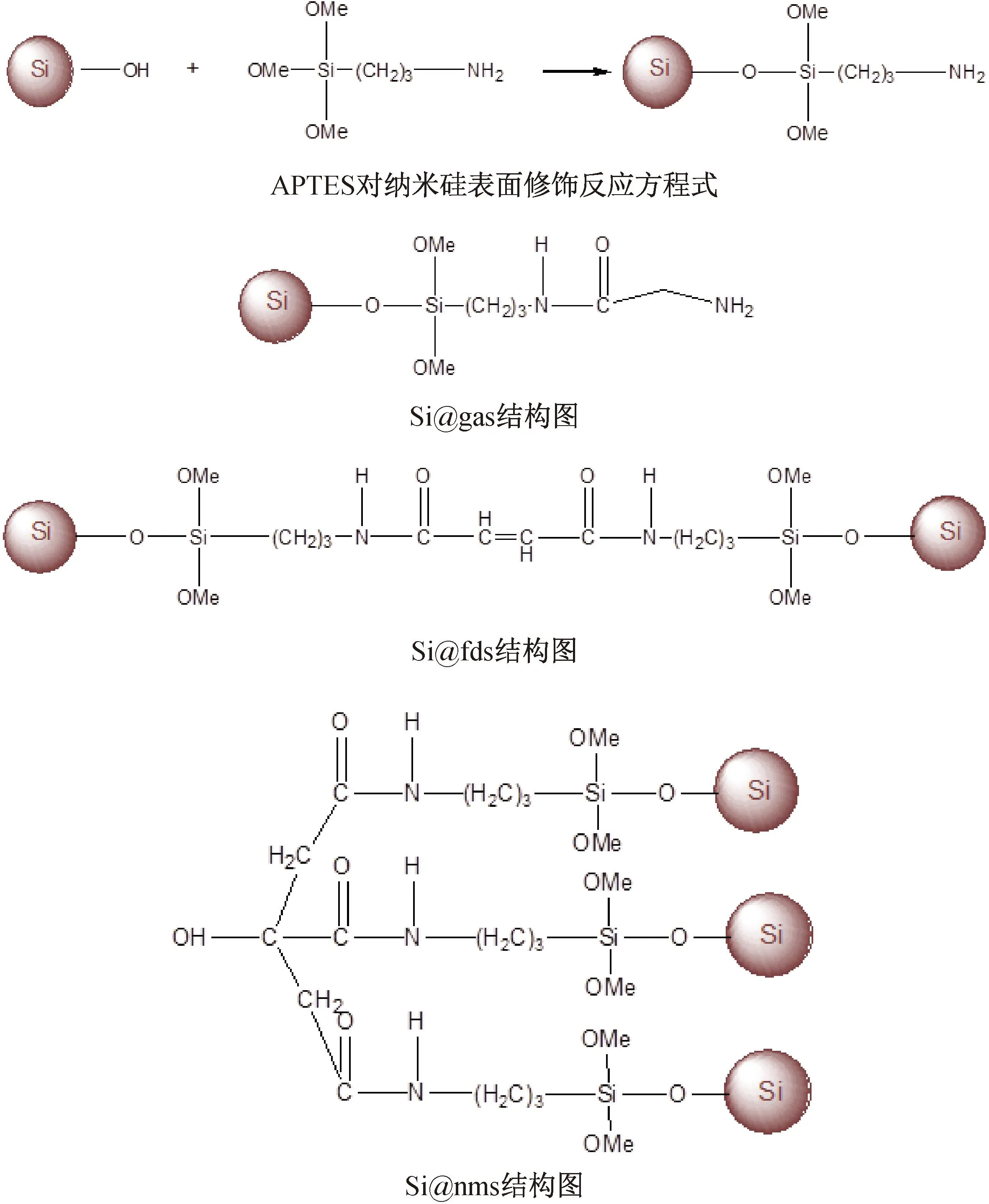
图2 APTES对纳米硅表面修饰反应方程式及与多元羧酸复合后的结构图
该合成材料的方式简单、低毒,对设备的需求低,且合成材料的形貌可以由参与反应的多元酸的结构变化所调控,是一种有效合理且充满前景的硅碳负极材料的制备方式。Si@C(nms)材料在提高硅碳材料比容量的同时,增加其循环稳定性,提升倍率性能,并且促进了锂离子扩散,降低了电化学阻抗。目前材料仅做的三元羧酸,希望可以通过进一步的探索,在各类不同结构多元羧酸及其衍生物的对比中,找到性能更加优异且成本更低的材料。
2.2 复合材料的表征分析
2.2.1 电镜分析 纳米硅粉和合成材料的扫描电镜图像如图3所示,纳米硅粉为粒径在100 nm左右的无定型颗粒,纳米硅粉复合甘氨酸,反丁烯二酸,柠檬酸之后,其中3种酸都与纳米硅粉结合起来。甘氨酸和反丁烯二酸的团聚现象更明显一些,尤其是甘氨酸,可能是由于该材料自身可以通过反应结合,导致分布的极不均匀。柠檬酸复合的纳米硅粉,分散的更加均匀,说明了纳米硅粉在柠檬酸溶液中分散的更好,更好的作为碳源复合在硅粉之上,在制备为极片时可以更好地形成导电网络,以及对纳米硅的膨胀进行更好的约束,维持电极结构。为了更好地说明这个问题,图4展示了对20%Si@C(nms)负极极片循环前和在500 mA/g电流密度下200圈循环后的极片横截面,循环前电极材料的厚度约为13.008 μm,经过循环后,电极材料的厚度约为16.880 μm,材料仅膨胀了23%,进一步说明该方法制备的材料对硅材料的束缚比较稳定。

图3 复合材料的扫描电镜图(a)纳米硅粉; (b)20%Si@C(gas);(c)20%Si@C(fds); (d) 20%Si@C(nms)

图4 20%Si@C(nms)组装电池循环前后的极片扫描电镜图(a)循环前; (b)循环后
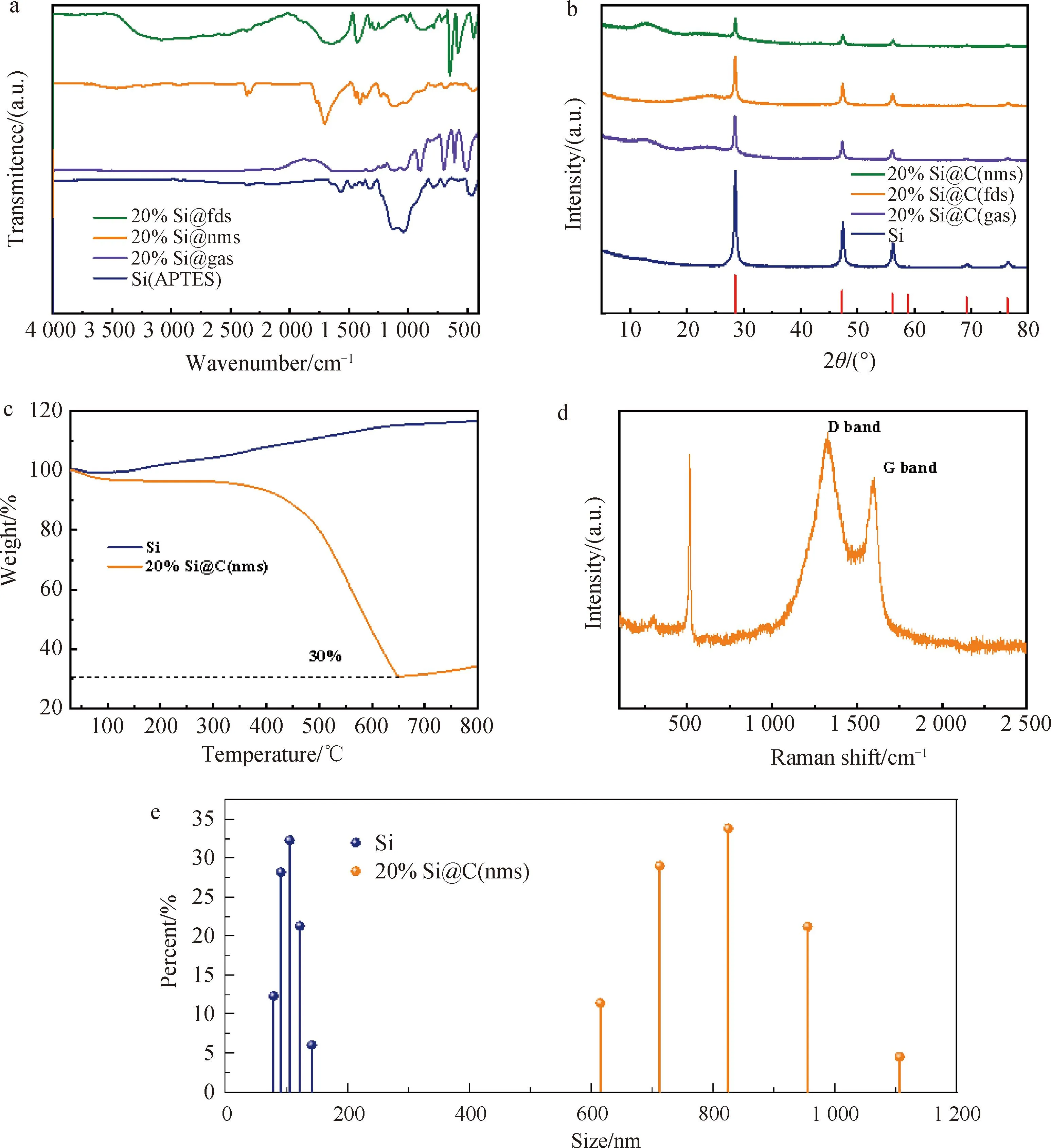
图5 (a)修饰后的纳米硅粉及3种材料的红外图谱,(b)纳米硅粉及3种材料的XRD图谱,(c)硅粉和20%Si@C(nms)的热重,(d)20%Si@C(nms)的拉曼光谱和(e)纳米硅粉和20%Si@C(nms)的粒径分布
修饰后的纳米硅粉及3种材料的XRD图谱如图5(b)所示,4种材料图谱中出现的28.3、47.3、56.0、69.1、76.4度分别对应于硅的(111)、(220)、(311)、(331)、(422)晶面[29]。XRD图谱中出现了纯硅的晶格面,因此4种材料中的硅结晶度好,结晶度明显。在3种材料的图谱中均出现了23°的宽峰,这是三种有机物碳化后产生碳材料特征峰碳[30],进一步说明3类复合材料的成功制备。
2.2.3 热重表征分析 在图5(c)的中,使用热重分析对纳米硅粉和20%Si@C(nms)的成分进行分析。在100 ℃以下,曲线有略微下降,可能由于材料中存在水分导致,在200 ℃后曲线逐渐上升,700 ℃后曲线停止上升到800 ℃一直趋于稳定。这个阶段的质量上升是由于纳米硅粉的氧化[31],由硅转化为二氧化硅所导致的质量上升。对于20%Si@C(nms)材料的热重曲线,前期100 ℃以下的质量损失也是由于水分的丧失,在650 ℃时材料质量损失约70%,这里主要是由于复合材料中碳材料被氧化去除导致的,但是同时在这个温度阶段也伴随着硅材料的氧化质量增长,所以判断出材料中的硅含量约在30%左右。之后650 ℃,曲线的上升对应的质量上升来自于未完全氧化的硅所贡献。
2.2.4 拉曼和粒径表征分析 20%Si@nms材料的拉曼光谱如图5(d)所示,对于碳材料,1 330、1 584 cm-1左右的峰分别代表D峰、G峰的典型特征,D峰和G峰的强度比可以反映材料的石墨化程度[32],20%Si@nms材料的ID/IG约为1.16,说明该材料石墨化程度较小,在拉曼图谱中还有一个510 cm-1的峰,该峰对应的是硅的峰。
图5(e)使用粒度分布仪对纳米硅粉和20%Si@C(nms)进行粒度分布测试,对于纳米硅材料,粒度分布在100 nm左右,小范围波动。对于20%Si@C(nms)材料,材料大小主要分布于800 nm,在700到900 nm之间进行波动。两者的粒径分布和二者的电镜图相互对应。由此也说明20%Si@C(nms)的颗粒单体是由纳米硅粉相互结合在一起所形成。
2.3 复合材料的电化学性能测试使用每种材料制备的电极极片和锂片制备成纽扣电池,进行一个电化学循环能力测试,如图6所示。在500 mA/g的电流密度下,Si@C(gas)材料的电化学性能由于复合的不均匀,该材料的循环特性在短周期内衰减到较低的容量,与纯硅材料有相似的循环特性。Si@C(fds)材料循环比较平稳,但是容量略微较低。对比之下可以看出20%Si@C(nms)材料的循环性能最佳,为此我们进一步对其电化学性能进行测试。
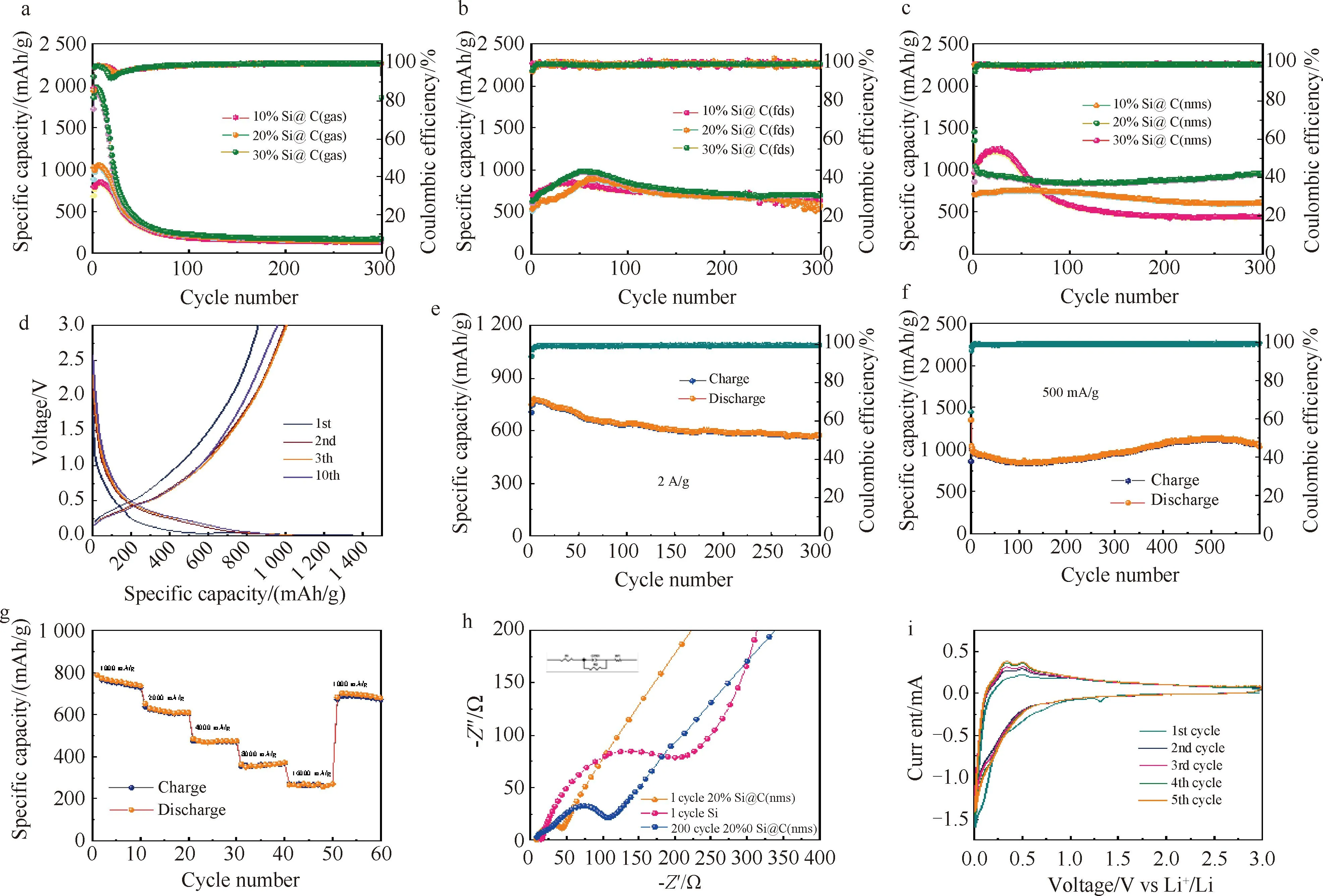
图6 (a)10%Si@C(gas), 20%Si@C(gas), 30%Si@C(gas)电池循环数据;(b)10%Si@C(fds), 20%Si@C(fds),30%Si@C(fds)电池循环数据; (c)10%Si@C(nms), 20%Si@C(nms), 30%Si@C(nms)电池循环数据;(d)20%Si@C(nms)的充放电曲线; (e)20%Si@C(nms)在2 A/g电流密度下循环性能; (f)20%Si@C(nms)在500 mA/g电流密度下长循环性能; (g)20%Si@C(nms)的倍率性能测试; (h)20%Si@C(nms)和硅的阻抗; (i)20%Si@C(nms)的CV
20%Si@C(nms)材料的长循环性能最佳,在500 mA/g的电流密度下,该材料初始充放电容量分别为1 350 mAh/g和861 mAh/g,初始库伦效率为63.7 %。其中的不可逆容量,主要是由于固体电解质膜(SEI)的形成。进一步地,我们对材料进行了大电流循环测试,在2 A/g的电流密度下,电池的初始充放电容量约为751 mAh/g和705 mAh/g,在循环几圈之后,电池的可逆比容量达到了770 mAh/g,电池容量略微提升,这是由于电池的活化导致的,在经过300圈的循环后,容量维持在573 mAh/g,在大电流的循环下容量衰减的幅度不大,进一步说明该材料的容量保持率优异。
在进行长循环测试时,20%Si@C(nms)材料在500 mA/g 的电流密度下进行700圈循环,电池容量维持于900 mAh/g和1 100 mAh/g之间,总体来说,材料的循环稳定性优异。相较与商用的硅碳负极材料650 mAh/g的容量有着一个显著的提升。在进行倍率测试时,初始的电流密度为1 000 mA/g,在这个电流密度下材料的比容量约为736 mAh/g,之后逐渐增加到16 000 mA/g,比容量下降到了270 mAh/g,当电流密度回到1 000 mA/g时,电池的比容量回到了约690 mAh/g,由此得出该材料的倍率性能较好。
对20%Si@C(nms)材料进行CV测试后,0.18 V为中心的还原峰是由晶态硅向非晶态LixSi的合金化反应引起的。而在0.36 V和0.54 V处的两个显著氧化峰属于LixSi的脱合金过程。不同扫描周期下的曲线面积变化不大,这表明20%Si@C(nms)电极材料的电化学稳定性较好。结果表明,该材料在循环的过程中,SEI膜的动态变化相于另外的材料更小,结构稳定性更强。为了进一步解释电极材料的电化学行为,对电池材料进行电化学阻抗测试。对比了纳米硅粉材料,20%Si@C(nms)第一圈阻抗以及200圈循环后的阻抗有着明显的下降,这说明有机碳源的复合能提升电极材料的导电能力,显著减低阻抗。
3 结论
本研究的出发点是由传统的成本较低的硅碳复合材料制备方法之一物理包覆出发,通过自组装的方式,为其增加了新的化学键合,同时选用尺寸更小的纳米硅粉减弱膨胀效应[33],2个策略对硅碳材料的稳定性进行改善,同时由于掺入的碳源在纳米硅之间相互胶黏,经过高温碳化之后,可以在不同纳米硅颗粒间形成碳导电网络。由于有机酸本身具有一定粘性,过量的有机酸材料会在纳米硅的外层以传统物理包覆的方式结合,也可以形成导电网络[34]。从两个方向出发,在保护硅电极的同时增加其导电性能。通过目前工作的探究可以发现该方式制备的20%Si@C(nms)材料具有出色的性能,在500 mA/g的电流密度下经过600圈的长循环也能保证在1 000 mA/g的比容量。同时该制备方式有着工艺简单,对设备要求低的优点,有利于实验向产业迈进。
猜你喜欢
黑龙江水利科技(2023年6期)2023-07-20
广西大学学报(自然科学版)(2019年1期)2019-03-18
山西建筑(2018年20期)2018-08-16
电镀与环保(2017年5期)2017-12-19
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年3期)2017-01-20
电镀与环保(2016年2期)2017-01-20
风能(2015年8期)2015-02-27
风能(2015年5期)2015-02-27
电测与仪表(2014年1期)2014-04-04
