
N3H3分子取代基效应的的量子化学研究
2014-10-09 03:39郭雅琼李强根
四川师范大学学报(自然科学版) 2014年5期
郭雅琼, 毛 双, 李强根
(1.四川师范大学化学与材料科学学院,四川成都610066; 2.武警警官学院,四川成都610213)
20世纪90年代后,由于高能量密度材料(HEDM)在军工、航天、能源等现代高科技工业领域中的重要意义[1-3],其合成研究及应用也受到世界各国的高度重视.实际使用的高能量密度材料一般都是由氧化剂、高能量密度化合物及其它添加剂构成的复合体系.由于氮氢化合物(NnHn)的不稳定性,因此在自然界中存在较少,大多数都以反应中间体或裂解产物的形式存在[4-5].但此类化合物在含能材料方面有重要作用,所以氮族化合物从20世纪50年代起就得到了重视和研究[6].近年来,关于氮氢化合物的研究越来越多,但由于其不稳定性,所以目前关于其研究仍大多集中在理论上.W.B.David[7-8]曾对N4H4和N6H6系列分子的几何构型构象变化和质子亲和势等方面进行了理论研究.文献也报道过采用G3B3方法研究了N4H4的几何构型、生成热、稳定性和互变异构现象[9].N3H3是氮族化合物中较为简单的一种物质,它和它的衍生物构成了一组很重要的化合物.通过对氮氢化合物的几何性质和能量的讨论,发现在氮氢化合物上引入其它基团后,分子的几何构型和能量将会发生变化.近几年来,课题组对NnHn(n=3~8)的某些氮氢化合物的结构与性质理论也进行了分析研究[10-12].在本文中主要考察环状和链状N3H3化合物中氢原子被甲基和羟基分别取代后,对原有分子的几何构型、能量所造成的影响.
1 计算方法
采用密度泛函方法(B3LYP)在6-311++G**基组下,对N3H3分子及其取代物进行了几何构型优化,并进行了振动分析.结果表明,计算所得到的构型均为势能面上的稳定点.然后,在此基组水平上运用自然键轨道(NBO)[13]分析方法对几何构型进行了NBO分析,揭示超共轭作用对取代物构型稳定性的影响.同时用AIM 2000程序包[14]对化合物的成键临界点电荷密度进行了分析,明确化学键的性质.此外采用G3MP2方法对分子能量进行校正,计算了这些异构体的生成热,所有计算都采用Gaussian 98程序[15].
2 结果与讨论
2.1 几何构型分析图1列出了环丙氮烷和丙氮烯的羟基异构体.在这里选择下列构型:1-羟基环丙氮烷(A)、1,2-二羟基环丙氮烷(D)和1,2,3-三羟基环丙氮烷(G),来讨论取代基的加入对环丙氮烷分子的影响.在这3个构型中H原子逐步被羟基(—OH)所取代.选择构型:1-羟基-1-丙氮烯(B)、1,3-二羟基-1-丙氮烯(E)和1,3,3-三羟基-1-丙氮烯(H)来讨论取代基对丙氮烯分子的影响.

图1分别列出了3种羟基(—OH)取代物的键长数据和AIM分析.从键长数据可以看出,N——N的键长范围在0.122 0 nm左右,环状构型的N—N键长大于链状构型,其中1,2-二羟基环丙氮烷(D)和1,2,3-三羟基环丙氮烷(G)N1—N2之间的键长大于0.150 0 nm,但经AIM分析发现其▽2ρ<0,表明所有的化学键仍为共价键.丙氮烯的H原子被羟基取代后,N——N双键的键临界点的电荷密度逐渐增大,而N—N单键的电荷密度逐渐减小,因此N——N双键的键长明显变短,N—N单键的键长明显增长.
2.2 能量分析表1列出了计算所得的各异构体的能量,其中E(NL)为超共轭作用能,E(L)为扣除超共轭作用后的分子能量.从表中数据可以看出,随着取代基数目的增加,总能量逐渐降低.所有异构体中一取代物有3种(A,B,C),二取代物有3种(D,E,F),三取代物有2种(G,H).对于一取代物A,B,C,其中B的分子能量最低,是最稳定的构型.二取代物中E是最稳定构型,三取代物H是最稳定的构型.分子的总能量与相对稳定性有关,而生成热是衡量高能材料爆炸性能的重要参数.采用G3MP2方法,在标压298 K下计算了各个异构体的生成热.异构体的总能量与生成热的变化趋势是一致的.当总能量越高,异构体的生成热就越大.为了进一步研究影响异构体稳定性的因素,采用NBO方法计算了它们的超共轭作用.通过比较发现,超共轭作用使体系能量降低,当扣除超共轭作用后,体系能量升高.在异构体中超共轭作用与羟基的个数成正比,随着羟基的个数的增加,分子的超共轭作用能逐渐增大.通过二阶稳定化能(E(2))分析,可以了解超共轭作用的实质.由表2可知,N3H3分子中的超共轭作用体现在N原子孤对电子与相邻的N—N键、N—H键之间.而H原子被羟基取代后,分子中就产生了N原子孤对电子与相邻的N—N键、N—H键、N—O键之间的相互作用,同时O原子与相邻的N—N键、N—O键之间也存在相互作用.
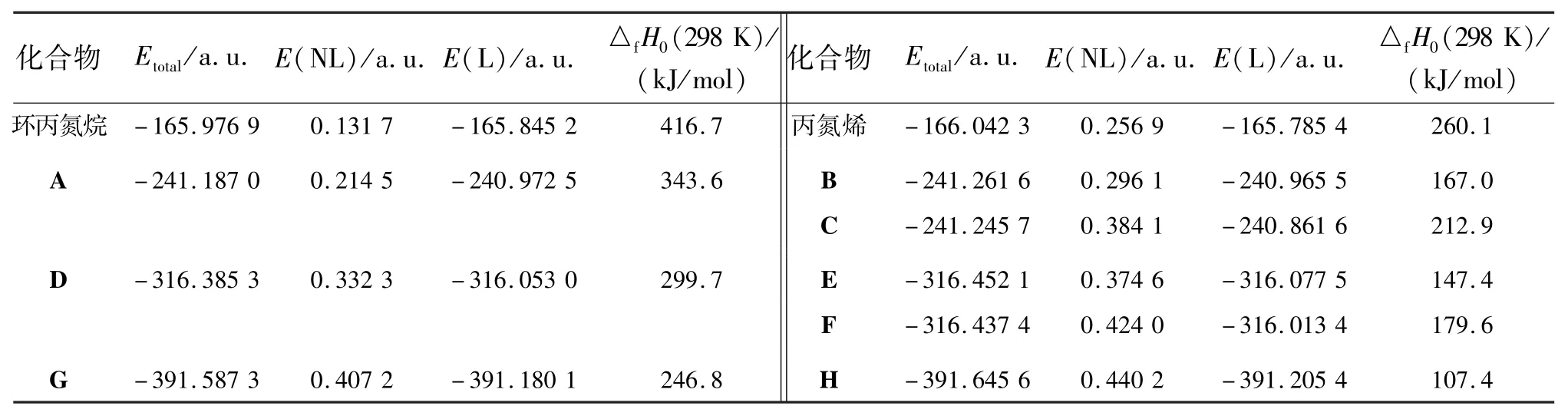
表1 化合物异构体的能量参数Table 1 Energy parameters calculated by various methods of conformers
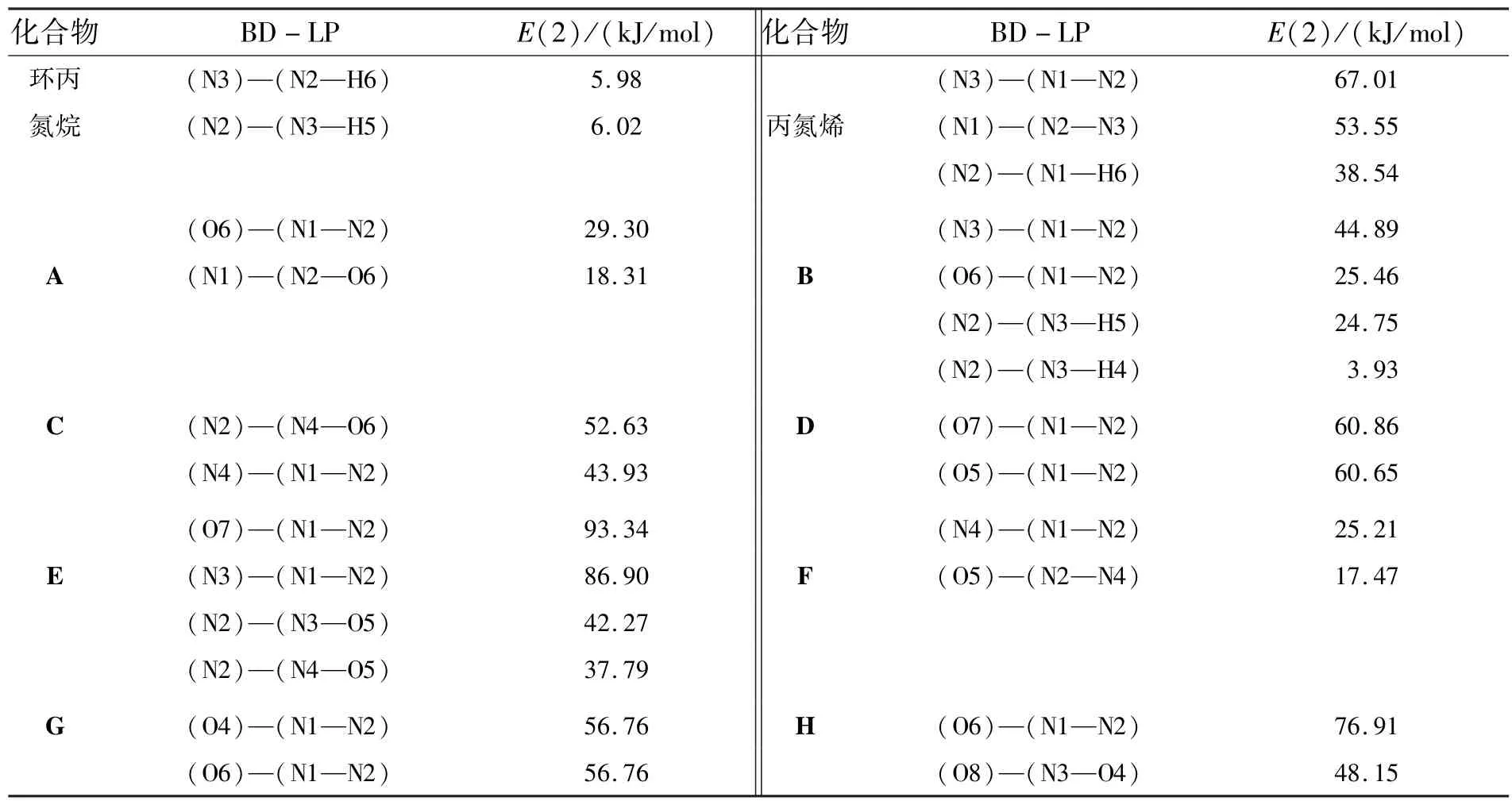
表2 化合物异构体的主要二阶稳定化能分析参数Table 2 Some significant second order perturbation energies of conformers
3 甲基(—CH3)的取代
3.1 几何构型分析图2列出了环丙氮烷和丙氮烯的甲基异构体.在这里选择下列构型:1-甲基环丙氮烷(I)、1,2-二甲基环丙氮烷(J)和1,2,3-三甲基环丙氮烷(K),来讨论取代基的加入对环丙氮烷分子的影响.在这3个构型中H原子逐步被甲基(—CH3)所取代.选择构型:1-甲基-1-丙氮烯(L)、1,3-二甲基-1-丙氮烯(N)和1,3,3-三甲基-1-丙氮烯(P)来讨论取代基对丙氮烯分子的影响.
图2分别列出了3种甲基(—CH3)取代物的键长数据和 AIM分析.从键长数据可以看出,N——N的键长范围在0.124 5 nm左右,环状构型的N—N键长范围在0.145 0 nm左右大于链状构型.经AIM分析发现,H原子被甲基(—CH3)取代后,氮氮键的性质没有改变,均为共价键(▽2ρ<0).丙氮烯的H原子被(—CH3)取代后,N——N双键的键临界点的电荷密度逐渐变小,而N—N单键的电荷密度增大,因此N——N双键的键长变长,N—N单键的键长变短.
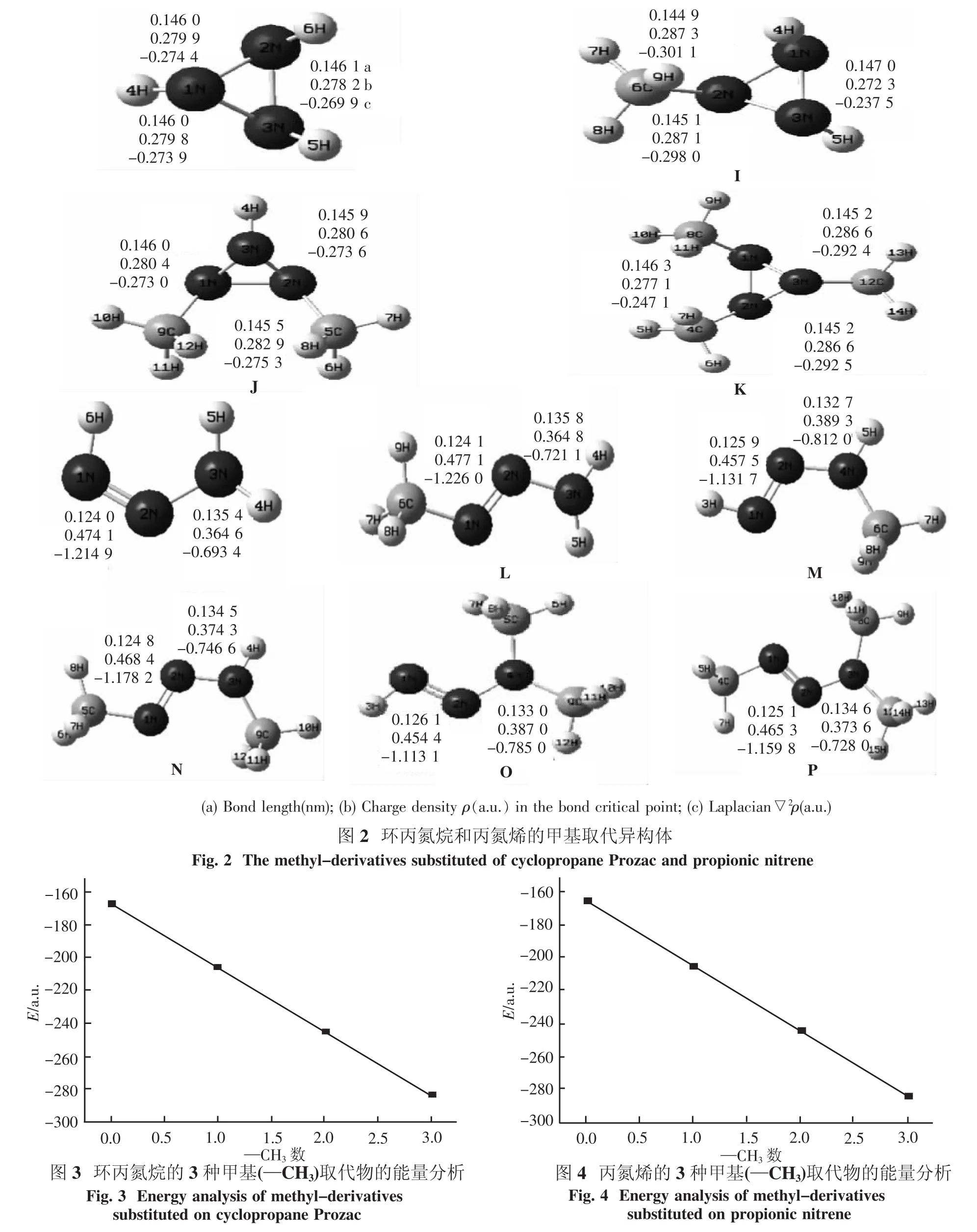
3.2 能量分析表3列出了计算所得的各异构体的能量.从表中数据可以看出,同样随着取代基数目的增加,总能量逐渐降低.所有异构体中一取代物有3种(I,L,M),二取代物有3种(J,N,O),三取代物有2种(K,P).对于一取代物中L是最稳定的构型,二取代物中N是最稳定构型,三取代物P是最稳定的构型.图3和图4分别列出了环丙氮烷和丙氮烯各个取代物的能量分析(在这里分别取L和N为丙氮烯分子的一取代物和二取代物作图).当H原子逐步被甲基(—CH3)取代后,整个分子的总能量会逐渐降低.以取代物分子中所含有的甲基数目对分子总能量作图,图3和图4中的横坐标表示分子中的甲基数目.从图上可以看出来,甲基数目对分子总能量都有很好的相关性.生成热也是考察含能物质的一个重要数据.分子的生成热均呈现降低的趋势.通过NBO计算,发现分子中的超共轭作用与甲基的个数成正比,随着甲基的个数增加,分子的离域化能逐渐增大.由表4可知当分子中的H原子逐渐被取代后,分子中就产生了N原子孤对电子与相邻的N—N键、N—C键、C—H键之间的相互作用.甲基(—CH3)取代所产生的超共轭作用能略高于羟基(—OH)取代的超共轭作用能.

表3 化合物异构体的能量参数Table 3 Energy parameters calculated by various methods of conformers

表4 化合物异构体的主要二阶稳定化能分析参数Table 4 Some significant second order perturbation energies of conformers
4 结论
本文主要讨论了2种N3H3的异构体(环丙氮烷和丙氮烯)中的氢原子被甲基和羟基取代后,对原有分子的几何构型、能量所造成的影响.通过几何构型优化和振动分析,表明所有异构体均为势能面上的稳定点.通过AIM分析,研究了化学键的本质,同时N—N键长与键临界点的电荷密度存在线性关系.经过NBO的超共轭作用计算发现,超共轭作用在决定构型稳定性的方面起了主要的作用.当引入甲基或羟基后,N原子的孤对电子会与相应的N—O(N—C)键之间发生相互作用,使整个分子的超共轭作用增强.随着取代基数目的增多,总能量和生成热都会降低,取代基数目与分子能量的降低值具有很好的相关性.
[1]Foltz M F,Holtz E V,Ornellas D O,et al.The solubilityo-fCL-20 in selected materials[J].Prop Expl Pyro,1994,19:206-212.
[2]Fried L E,Manaa M R,Pagoria P F,et al.Design and synthes is ofenergeticm at erials[J].Mater Res,2001,31:291-295.
[3]阳世清,徐松林,黄亨健,等.高氮化合物及其含能材料[J].化学进展,2008,20(4):526-537.
[4]舒远杰.含能材料:辉煌的20世纪及其前途[C]//四川省中青年专家大会论文集.绵阳:中国工程物理研究院出版社,2002:10.
[5]Li Y C,Cai Q,Li S H,et al.1,1'-Azobis-1,2,3-triazole:a high-nitrogen compound with stable N8structure and photochromism[J].J Am Chem Soc,2010,132(35):12172-12173.
[6]Giguère P A,Liu I D.On the infraredspectrum of hydrazine[J].J Chem Phys,1952,20:136-140.
[7]David W B.Thermochemical properties of diaziridine 1,2-diazetidine and 1,3-diazetidine[J].J Mol Struct,2005,730:95-103.
[8]David W B.Hartree Fock Gaussian-2 and-3 and complete basis set predictions of some thermochemical properties of N4H6[J].J Phys Chem,2001,105:465-470.
[9]毛双,李来才.N4H4分子取代基效应的量子化学研究[J].原子与分子物理学报,2012,29(5):1386.
[10]毛双,谭英雄,蒲雪梅,等.N5H5的结构性能的量子化学研究[J].四川大学学报:自然科学版,2009,46(4):1089.
[11]Mao S,Pu X M,Li L C,et al.Theoretic study on the structure and property of N6H6[J].Acta Chim,2006,64(14):1429.
[12]Mao S,Pu X M,Li L C,et al.Theoretical predictive study on N7H7hydronitrogen compounds[J].J Mol Struct Theochem,2008,858:12.
[13]Reed A E,Weinhold F,Curtiss L A,et al.Natural bond orbital a nalysis of molecular interactions:theoretical studies of binary complexes of HF,H2O,NH3,N2,O2,F2,CO and CO2with HF,H2O and NH3[J].Chem Phys,1986,84:5687-5706.
[14]Biegler K F,Schonbohm J,Derdan R,et al.AIM 2000[S].Ver 2.0.Hamilton:McMaster University,2000.
[15]Frisch M J,Truck G W,Schlegel H B,et al.Gaussian 98[S].Rev A.02.Pittsburgh PA:Gaussian,1998.
猜你喜欢
系统仿真技术(2022年4期)2023-01-17
数学物理学报(2022年1期)2022-03-16
云南化工(2021年8期)2021-12-21
数学物理学报(2021年6期)2021-12-21
河北理科教学研究(2020年1期)2020-07-24
应用数学(2020年2期)2020-06-24
青岛大学学报(工程技术版)(2019年2期)2019-09-10
国外医药(抗生素分册)(2016年4期)2016-07-12
信息记录材料(2016年4期)2016-03-11
枣庄学院学报(2015年5期)2016-01-09
