
液相色谱-四极杆飞行时间质谱检测染毒动物血浆中沙林和梭曼酪氨酸加合物
2015-04-18 03:47于惠兰裴承新刘石磊
质谱学报 2015年3期
于惠兰,裴承新,胡 真,刘石磊,向 宇
(1.国民核生化灾害防护国家重点实验室, 北京 102205;2.防化研究院, 北京 102205)
液相色谱-四极杆飞行时间质谱检测染毒动物血浆中沙林和梭曼酪氨酸加合物
于惠兰1,2,裴承新1,2,胡 真2,刘石磊1,2,向 宇2
(1.国民核生化灾害防护国家重点实验室, 北京 102205;2.防化研究院, 北京 102205)
为了追溯性检测神经性毒剂沙林(GB)和梭曼(GD)暴露染毒,以染毒白蛋白上的特异性加合位点-411位酪氨酸加合物为重要生物标志物,建立了高效液相色谱-四极杆飞行时间质谱(LC/Q-TOF MS)分析动物血液中神经性毒剂GB和GD酪氨酸加合物的检测方法。在采用不同样品处理和仪器条件的基础上,确定了最佳实验条件。血液经离心后,取50 μL分离的血浆加入到50 μL 10 g/L链霉蛋白酶的NH4HCO3(50 mmol/L)水溶液中,于37 ℃孵育2.5 h,用0.5 mL 10 ku超滤管以16 000 r/min离心15 min,LC/Q-TOF MS电喷雾正离子模式分析滤液。应用本方法进行GB半致死剂量(LD50)动物施毒实验,在施毒的动物血样中检测到膦酰化酪氨酸加合物,证明了该方法快速、简单、可靠。
生物标志物;神经性毒剂;沙林(GB);梭曼(GD);血浆;酪氨酸加合物;液相色谱-四极杆飞行时间质谱(LC/Q-TOF MS)
自第一次世界大战以来,化学毒剂(chemical warfare agents,CWAs)作为大规模杀伤性武器被多次使用,其中,神经性毒剂的毒性、杀伤力最强,可在μg/kg剂量范围内造成死亡。典型的神经性毒剂有G类和V类两种,G类毒剂的代表是沙林(GB)和梭曼(GD),而V类毒剂的代表是维埃克斯(VX),其结构式示于图1。
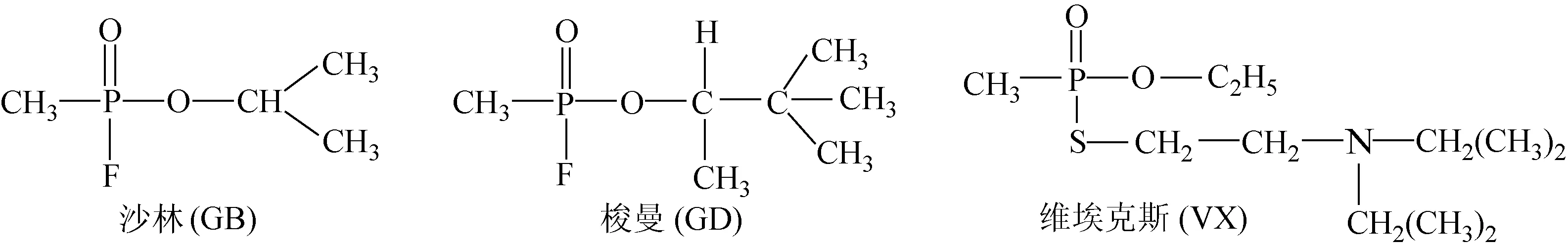
图1 典型的神经性毒剂的化学结构式Fig.1 Structures of typical nerve agents
多年来,禁止化学武器公约组织(OPCW)一直计划将生物医学样品的分析纳入化学武器核查指定的实验室分析网络。与环境基质样品相比,毒剂在生物体内存在的时间较长,具有较强的溯源性,因此,对生物医学样品的分析检测可以提供生物体接触神经性毒剂的明确证据。虽然白蛋白和毒剂的结合常数远小于胆碱酯酶与神经性毒剂的结合常数[1-5],但是生物体内白蛋白的浓度是胆碱酯酶浓度的1 000倍,同时白蛋白与毒剂加合物具有较长的半衰期,P—O官能团上的烷基不容易脱去,即不易发生衰变,是一类溯源性较强的生物标志物,有助于明确鉴定染毒的神经性毒剂种类[1-6]。
目前,在生物医学样品分析中,液相色谱-质谱(LC/MS)是首选技术[7-16],主要原因是相对于气相色谱-质谱(GC/MS)技术而言,LC/MS分析的是生物标志物本身,样品不用衍生等处理步骤,加快了分析进程,减少了试剂对生物标志物的干扰,而GC/MS是通过还原反应将染毒生成的加合物和(或)代谢物重新还原成毒剂原体[17-19]进行分析,不能明确鉴定每一种生物标志物。LC/MS通常采用液相色谱-四极杆飞行时间质谱(LC/Q-TOF MS)和液相色谱-三重四极杆质谱(LC-QQQ MS)两种技术。LC/Q-TOF MS是一种四极杆和飞行时间质谱相结合的具有高分辨功能(质量数精确到小数点后第3位)的技术,对于未知的生物标志物,可以获得MS/MS碎片离子的精确质量数,排除样品中可能的共流出基质干扰物对生物标志物的干扰,降低化学噪音,从而提高定性结果的可靠性。LC-QQQ MS技术主要是对已知化合物进行分析,属于单位质量分辨质谱技术,采用仪器中的多反应监测模式(MRM)对化合物进行鉴定,灵敏度比LC/Q-TOF MS高。
为了能够鉴定未知的生物标志物,本研究采用LC/Q-TOF MS技术检测经链霉蛋白酶酶解后的GB和GD染毒的动物血浆中生物标志物——膦酰化酪氨酸加合物,并通过活体动物GB施毒实验验证,以期建立适合染毒生物医学样品的高通量分析方法。
1 实验部分
1.1 仪器与器材
Agilent HPLC 1200/6520 Q-TOF高效液相色谱/四极杆-飞行时间质谱仪:美国Agilent公司产品;SPE固相萃取仪:日本GL Sciences 公司产品;Anke TGL-16G离心机:上海安亭科学仪器厂产品;HB-202 Thermo cell恒温金属浴槽:杭州博日科技有限公司产品;10 ku超滤管:美国Millipore公司产品;移液枪:北京吉尔森公司产品;ProteoExtract®白蛋白去除试剂盒:德国Merk公司产品。
1.2 试剂
乙腈(HPLC级):美国J&K公司产品;纯净水:杭州娃哈哈公司产品;甲酸(LC/MS级):美国Agilent公司产品;白蛋白结合缓冲液(250 mmol/L磷酸钠,pH 7.4)、白蛋白去除缓冲液(25 mmol/L磷酸钠,pH 8,2 mol/L NaCl)、碳酸氢铵(纯度98%):均为比利时Acros公司产品;链霉蛋白酶:瑞士Roche公司产品;乙二胺四乙酸钠(EDTA,分析纯):北京化工厂产品。
1.3 血液与GB和GD孵育及酶解
1.3.1 血液与GB和GD孵育 从若干只比格狗和大耳白兔静脉各取1 mL全血,置于含一定量乙二胺四乙酸的5 mL塑料离心管中,加入一定量的GB、GD乙腈溶液,在37 ℃孵化,轻轻晃动,2.5 h后取出,以16 000 r/min离心3 min,将染毒全血分离成红细胞和血浆,取出一定量血浆,剩余部分于-20 ℃下冷冻,备用。
1.3.2 链霉蛋白酶酶解血浆 取50 μL染毒血浆,加入等体积的50 mmol/L NH4HCO3水溶液稀释,然后再加入50 μL 10 g/L链霉蛋白酶的NH4HCO3水溶液,于37 ℃孵化2.5 h,用0.5 mL 10 ku超滤管以16 000 r/min高速离心15 min,用100 μL移液枪取出试管中的小分子液体置于装有200 μL内衬管的自动进样器小瓶中,供LC/MS分析。
1.4 液相色谱及质谱条件
色谱柱:Agilent zorbax Eclipse plus C18柱(2.1 mm×150 mm×5 μm);流动相:水(0.05% 甲酸)(A)-乙腈(B)体系;梯度条件:5%B(0~2 min)—50%B(2.5 min)—100%B(6~12 min)—5%B(13~17 min);流速0.25 mL/min;进样体积5 μL;柱温30 ℃。
电离方式:电喷雾(ESI)离子源;正离子扫描;全扫描方式,小分子扫描范围m/z50~1 000,大分子扫描范围m/z50~3 000,扫描时间0.77 s,分辨率大于10 000;毛细管电压3.5 kV;气体温度350 ℃;干燥气(氮气)流速8 L/min;雾化气(氮气)压力2.07×105Pa;碎裂电压150 V;MS/MS碰撞能量20 V。
2 结果与讨论
2.1 色谱条件的选择
为了避免毒剂的水解与醇解,将毒剂配制在乙腈中,部分实验采用乙腈作为沉淀剂进行样品处理,为了保证溶剂的一致性,选择乙腈和水作为流动相,在不同的梯度条件下洗脱,其洗脱条件列于表1。实验发现,乙腈含量高,目标加合物的信噪比高,即检测灵敏度高;但是由于血浆样品比较复杂,如果乙腈含量过高,有可能使杂质等干扰物和目标物分离不完全。经实验验证,采用梯度洗脱条件4,在分析时间较短的情况下,能够保证加合物与干扰物有很好的分离,且灵敏度较高,因此最佳的梯度洗脱条件为条件4。
2.2 质谱条件的选择
为了使目标物获得较高的响应值,对质谱条件进行了优化。采用高分辨ESI正离子模式,m/z50~1 000的全扫描模式扫描,有效地提高了目标化合物信号的信噪比。由于检测的是超痕量目标化合物,考虑到灵敏度,所以优化了采集质谱图的频率(1.1、1.3、1.5 spectra/s)。由于采用的水相比例较大,为了最大限度的降低质谱信号中的噪音,优化了干燥气流量(5、8、10 L/min)、喷雾压力(1.38×105、2.07×105、2.76×105Pa)、干燥气流温度(250、300、350 ℃)、毛细管电压(3.2、3.5 kV)和碎裂电压(100、120、150 V)等5个主要指标。最终确定最佳质谱条件为:干燥气流量8 L/min,喷雾压力2.07×105Pa,干燥气流温度350 ℃,ESI+毛细管电压3.5 kV,碎裂电压150 V,采集谱图的频率1.3 spectra/s。
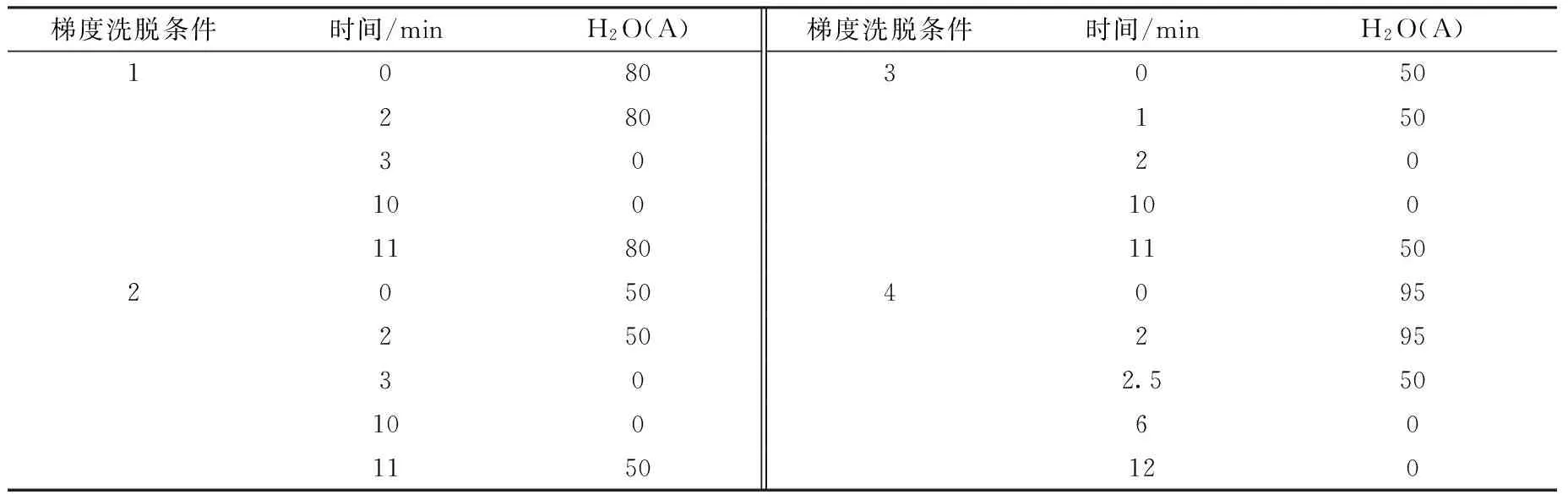
表1 GB和GD染毒酪氨酸加合物的梯度洗脱条件
2.3 白蛋白分离
采用白蛋白去除试剂盒分离纯化白蛋白。首先取180 μL分离的血浆,采用10倍结合缓冲剂稀释样品,即将180 μL血浆加入到180 μL 10倍结合缓冲剂中,然后加入1 440 μL高纯水。白蛋白试剂盒的处理方法为:采用高纯水将适当的10倍白蛋白结合缓冲液稀释成1倍结合缓冲剂,将6~10 mL 1倍结合缓冲剂用一次性注射器以0.1 mL/min的流速注入白蛋白去除小柱,用来平衡小柱,去除小柱内的空气;将稀释的样品用新的注射器以每5秒2滴的流速注入平衡后的小柱,使样品中的白蛋白与小柱上的键合相充分结合;用2 mL 1倍结合缓冲剂淋洗小柱,用新的一次性注射器取2 mL白蛋白去除缓冲剂,以0.1 mL/min的流速洗脱小柱上的白蛋白,收集流出液;将收集的流出液用10 ku超滤管以16 000 r/min超滤10 min,去除其中的小分子,用高纯水淋洗样品3次,分别以16 000 r/min超滤10 min,去除缓冲剂。留在超滤管中浓缩的大分子样品共计150 μL,用200 μL移液枪取出置于带有200 μL内衬管的自动进样器小瓶中,备用。
与直接将血浆酶解的方法比较,白蛋白去除试剂盒样品处理方法比较复杂,所得的染毒白蛋白分子质量与未染毒分子质量没有明显的偏差,酶解后检测到的酪氨酸加合物与血浆直接酶解方法检测到的酪氨酸加合物基本一致,因此可采用直接将血浆酶解超滤后分析。
2.4 LC/MS分析
经链霉蛋白酶消解后的GB染毒血样,通过LC/MS和LC-MS/MS鉴定得到膦酰化酪氨酸加合物MeP(O)(OiPr)-Tyr,经LC-MS/MS分析发现,质谱图中有m/z97甲基膦酸特征峰。链霉蛋白酶消解的未染毒样品、GB染毒样品和标样的ESI+-MS提取离子色谱图(EIC)及标样的MS/MS串联质谱图示于图2。按照同样方法分析链霉蛋白酶消解的未染毒样品、GD染毒样品和标样的ESI+-MS EIC图及标样的MS/MS串联质谱图,示于图3。

注:a. 空白;b. 样品;c. 标样;d. GB酪氨酸加合物标样图2 GB染毒膦酰化酪氨酸加合物的ESI+-MS EIC图及标样的ESI+-MS/MS图Fig.2 ESI+-MS EIC and MS/MS spectrum of phosphylated tyrosine adduct exposure to GB
2.5 LC-MS/MS谱图解析
对于GB染毒膦酰化酪氨酸加合物,MS谱图中基峰为准分子离子峰[M+H]+m/z302.116 2,计算的元素组成为C13H21NPO5;MS/MS质谱图中基峰为m/z214.063 0,计算的元素组成为C9H13NPO3。其他碎片离子为m/z260.068 7、197.036 2、136.075 3、118.065 0、97.005 8,碰撞电压20 V,几乎不见准分子离子峰。在一定电压下,异丙基官能团容易脱去,形成m/z260.068 7碎片离子,计算的元素组成为C10H15NPO5;进一步失去酪氨酸上的羧基,形成m/z214.063 0碎片离子;同时质谱图中的m/z197.036 2碎片离子是由m/z214.063 0碎片离子失去NH3形成的,计算的元素组成为C9H10PO3;m/z136.075 3计算的元素组成为C8H10NO;m/z118.065 0计算的元素组成为C8H8N;其中甲基膦酸特征峰m/z97.005 8计算的元素组成为CH5PO3,其裂解途径示于图4。对于GD染毒膦酰化酪氨酸加合物,MS谱图中基峰为m/z260.069 1碎片离子,m/z366.144 7是[M+Na]+峰。在一定碰撞电压下,GD染毒膦酰化官能团很快衰变,P—O官能团上的频哪基很容易脱去,与文献报道一致[8,10],其余裂解规律同GB染毒膦酰化酪氨酸加合物。
2.6 动物染毒实验应用研究
选取3只(2.5±0.05)kg大耳白兔,分别静脉注射半致死剂量的新鲜配制的GB20%乙醇水溶液。注射后,大耳白兔均有全身震颤、呼吸困难、瞳孔缩小等神经性毒剂中毒症状,3只白兔的染毒剂量分别为20、22、22 μg/kg。在染毒1 h、3 h、6 h、1 d、2 d、5 d和8 d共7个时间段,分别抽取静脉血,分析血浆中是否含有白蛋白膦酰化酪氨酸加合物。实验表明,在兔染毒5 d内,没有检测到GB与酪氨酸加合物,但在兔染毒8 d后,抽取的3个血样样品中均发现了少量的膦酰化酪氨酸,其ESI+-MS EIS图示于图5。

注:a. 空白;b. 样品;c. 标样;d. GD酪氨酸加合物标样图3 GD染毒膦酰化酪氨酸加合物的ESI+-MS EIC图及标样的ESI+-MS/MS图Fig.3 ESI+-MS EIC and MS/MS spectrum of phosphylated tyrosine adduct exposure to GD

图4 GB膦酰化酪氨酸加合物的ESI+-MS/MS质谱裂解途径Fig.4 ESI+-MS/MS fragmentation pathway of phosphylated tyrosine adduct exposure to GB

注:a. 空白; b. 1#兔; c. 2#兔; d. 3#兔; e. 沙林酪氨酸加合物标样图5 GB染毒样品中膦酰化酪氨酸的ESI+-MS EIC图Fig.5 ESI+-MS EIC of phosphylated tyrosine adduct exposure to GB in rabbit plasma
3 结论
通过白蛋白提取、纯化和链霉蛋白酶酶解等样品处理方法,建立了神经性毒剂GB和GD染毒白蛋白生物标志物——膦酰化酪氨酸的LC/Q-TOF MS和MS/MS定性方法,经GB染毒动物活体实验验证,该方法具有普适性,是一种较理想的神经性毒剂染毒溯源性检测方法。
[1] FIDDER A, NOORT D, HULST A G, et al. Retrospective detection of exposure to organophosphorus anti-choline sterases: Mass spectrometric analysis of phosphylated human butyrylcholinesterase[J]. Chem Res Toxicol, 2002, 15(4): 582-590.
[2] NOORT D, FIDDER A, van der SCHANS M J, et al. Verification of exposure to organophosphates: Generic mass spectrometric method for detection of human butyrylcholinesterase adducts[J]. Anal Chem, 2006, 78(18): 6 640-6 644.
[3] TSUGE K, SETO Y. Detection of human butyrylcholinesterase nerve gas adducts by liquid chromatography-mass spectrometric analysis after in gel chymotrytic digestion[J]. J Chromatogr B, 2006, 838(1): 21-30.
[4] SUN J C, LYNN B C. Development of a MALDI-TOF-MS method to identify and quantify butyrylcholinesterase inhibition resulting from exposure to organophosphate and carbamate pesticides[J]. J Am Soc Mass Spectrom, 2007, 18(4): 698-706.
[5] LI H, SCHOPFER L M, NACHON F, et al. Aging pathways for organophosphate-inhibited human butyrylcholinesterase, including novel pathways for isomalathion, resolved by mass spectrometry[J]. Toxicol Sci, 2007, 100 (1): 136-145.
[6] BLACK R M. History and perspectives of bioanalytical methods for chemical warfare agent detection[J]. Journal of Chromatography B, 2010, 878(17/18): 1 207-1 215.
[7] BLACK R M, HARRISOM J M, READ R W. The interaction of sarin and soman with plasma proteins: The identification of a novel phosphylation site[J]. Arch Toxicol, 1999, 73(2): 123-126.
[8] PEEPLES E S, SCHOPFER L M, DUYSEN E G. Albumin, a new biomarker of organophosphorus toxicant exposure, identified by mass spectrometry[J]. Toxicol Sci, 2005, 83(2): 303-312.
[9] LI B, SCHOPFER L M, HINRICHS S H, et al. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry assay for organophosphorus toxicants bound to human albumin at Tyr411[J]. Anal Biochem, 2007, 361(2): 263-272.
[10]WILLIAMS NH, HARRISON J M, READ R W, et al. Phosphylated tyrosine in albumin as a biomarker of exposure to organophosphorus nerve agents[J]. Arch Toxicol, 2007, 81(9): 627-639.
[11]BLACK R M. An overview of biological markers of exposure to chemical warfare agents[J]. Journal of Analytical Toxicology, 2008, 32(1): 2-9.
[12]READ R W, HARRISON J M,STEVENS J A, et al. Biomarkers of organophosphorus nerve agent exposure: Comparison of phosphylated butyrylcholinesterase and phosphylated albumin after oxime therapy[J]. Arch Toxicol, 2010, 84(1): 25-36.
[13]JOHN H, BREYER F, THUMFART J O, et al. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) for detection and identification of albumin phosphylation by organophosphorus pesticides and G- and V-type nerve agents[J]. Anal Bioanal Chem, 2010, 398(6): 2 677-2 691.
[14]BAO Y, LIU Q, CHEN J, et al. Quantification of nerve agent adducts with albumin in rat plasma using liquid chromatography-isotope dilution tandem mass spectrometry[J]. Journal of Chromatography A, 2012, 1 229: 164-171.
[15]BLACK R M, READ R W. Biological markers of exposure to organophosphorus nerve agents[J]. Arch Toxicol, 2013, 87(3): 421-437.
[16]CHEN S G, ZHANG J, LUMLEY L, et al. Immunodetection of ierum albumin adducts as biomarkers for organophosphorus exposures[J]. J Pharmacol Exp Ther, 2013, 344(2): 531-541.
[17]NOORT D, HULST A G, PLATENBURG D H J M, et al. Quantitative analysis of O-isopropyl methylphosphonic acid in serum samples of Japanese citizens allegedly exposed to sarin: Estimation of internal dosage[J]. Arch Toxicol, 1998, 72(10): 671-675.
[18]NOORT D, BLACK R M. Methods for the retrospective detection of exposure to toxic scheduled chemical. Part B. Mass spectrometry and immunochemical analysis of covalent adducts to proteins and DNA. In: Mesilaakso M(ed) Chemical weapons convention chemicals analysis(sample collection, preparation and analytical methods)[M]. West Sussex: Wiley, 2005: 403-431.
[19]ADAMS T K, CAPACIO B R, SMITH J R. The application of the fluoride reactivation process to the detection of sarin and soman nerve agent exposures in biological samples[J]. Drug Chem Toxicol, 2004, 27(1): 77-91.
Determination of Sarin and Soman Tyrosine Adducts in Exposed Animal Plasma by Liquid Chromatography/Quadrupole-Time of Flight Mass Spectrometry
YU Hui-lan1,2, PEI Cheng-xin1,2, HU Zhen2, LIU Shi-lei1,2, XIANG Yu2
(1.StateKeyLaboratoryofNBCProtectionforCivilian,Beijing102205,China;2.ResearchInstituteofChemicalDefence,Beijing102205,China)
In order to retrospective analysis of biomedical samples exposed to nerve agents sarin (GB) and soman (GD), a method for determination of GB tyrosine adducts and GD tyrosine adducts in exposed animal plasma as a biomarker was developed by liquid chromatography/quadrupole-time of flight mass spectrometry(LC/Q-TOF MS). After optimizing different sample preparation processes and a series of instrumental parameters, the adducts were analyzed by LC/Q-TOF MS in positive electrospray ionization mode. After centrifugation, 50 μL plasma was added into 50 μL 10 g/L Pronase E in ammonium bicarbonate(50 mmol/L)and incubating at 37 ℃ for 2.5 h. The supernatant was analyzed after filtered through 0.5 mL molecular mass cutoff filters (10 ku) under centrifugation at 16 000 r/min for 15 min. The method is rapid, simple and reliable, which is suitable for analyzing the phosphylated tyrosine adducts in blood samples from the rabbits injected with LD50of GB.
biomarker; nerve agent; sarin(GB); soman(GD); plasma; tyrosine adduct; liquid chromatography/quadrupole -time of flight mass spectrometry(LC/Q-TOF MS)
2014-05-07;
2014-08-21
于惠兰(1972—),女(汉族),山东文登人,副研究员,从事军事化学研究。E-mail: yuhuilan1@163.com
时间:2015-01-30;
http:∥www.cnki.net/kcms/detail/11.2979.TH.20150130.1515.004.html
O657.63
A
1004-2997(2015)03-0261-07
10.7538/zpxb.youxian.2015.0002
猜你喜欢
理化检验-化学分册(2022年11期)2022-11-27
食管疾病(2022年1期)2022-11-26
动物营养学报(2022年3期)2022-03-30
化工管理(2021年4期)2021-02-27
——第二部分:原棉短纤维率标样的验证试验分析
中国纤检(2020年7期)2020-07-22
天然产物研究与开发(2019年8期)2019-09-05
环球时报(2018-04-16)2018-04-16
医学研究杂志(2015年9期)2015-07-01
城市建设理论研究(2012年4期)2012-03-23
军事历史(1993年2期)1993-01-18
