
壮腰健肾小蜜丸的质量标准研究*
2015-04-25 08:08河北省沧县疾病预防控制中心牛秀丽张天鹏沧州061000
河北中医药学报 2015年2期
河北省沧县疾病预防控制中心 牛秀丽 张天鹏 石 红 张 旺(沧州 061000)
壮腰健肾小蜜丸是由狗脊、鸡血藤、金樱子、黑老虎、牛大力、桑寄生、菟丝子、千斤拨、女贞子9味中药组成,具有壮腰健肾,养血,祛风湿的功能。一般常用于肾亏腰痛,膝软无力,小便频数,风湿骨痛,神经衰弱。原标准无薄层鉴别方法和高效液相色谱法,参照壮腰健肾片质量标准,[1]制定了壮腰健肾小蜜丸质量标准。
1 实验仪器与材料
1.1 仪器 层析缸 (上海信谊仪器厂);微量点样毛细管 (华西医大科学仪器公司);薄层层析硅胶G(龙海药业有限责任公司自制);GH电热恒温培养箱 (天津市泰斯特仪器有限公司);TGL-16C型台式高速离心机 (上海安亭科学仪器厂制造);QT10260超声波清洗机 (天津市瑞普电子仪器公司);ZF-I型三用紫外分析仪 (上海顾村电光仪器厂)。
1.2 材料 硅胶G(青岛海洋化工集团);牛大力对照药材 (批号:112687-200901中检所);黑老虎对照药材 (批号:121438-201001中检所);鸡血藤对照药材 (批号:121173-201003中检所);金樱子对照药材 (批号:121173-201003中检所);原儿茶酸对照品 (批号:110809-200903中检所);壮腰健肾小蜜丸 (某公司生产,批号:110302、110303、110304);其余试剂为分析纯。
2 定性鉴别
2.1 黑老虎、牛大力和鸡血藤鉴别[1-2]取小蜜丸 (批号:110302)15 g,剪碎,加硅藻土3 g,研匀,置具塞锥形瓶中,加水20 mL,润湿,再加乙酸乙酯-甲醇 (4︰1)100 mL,超声处理30 min,滤过,滤液蒸干,残渣加甲醇2 mL使溶解,作为供试品溶液。另分别取黑老虎、牛大力和鸡血藤对照药材各1.5 g,置锥形瓶中,加水10 mL润湿,再加乙酸乙酯-甲醇 (4︰1)50mL,按照供试品制备方法制成对照药材溶液。照薄层色谱法(中国药典2010版一部附录VIB)试验,吸取供试品溶液10μL,黑老虎、牛大力和鸡血藤对照药材溶液各5μL,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以甲苯-乙酸乙酯--甲酸 (10︰7︰1)为展开剂,展开,取出,晾干,置紫外灯 (365 nm)下检视。供试品色谱中,在与黑老虎、牛大力和鸡血藤对照药材色谱相应位置上,显相同颜色的荧光斑点。
2.2 金樱子鉴别[3-4]取金樱子对照药材3 g,加水100 mL煎煮1 h,冷却后,过滤,滤液离心,收集离心液,加乙酸乙酯液萃取2次,每次30 mL,合并乙酸乙酯层溶液,蒸干,残渣加1 mL甲醇使溶解,即为对照药材溶液。照薄层色谱法 (中国药典2010版一部附录VI B)试验,吸取〔鉴别〕2.1项下的供试品溶液及上述对照药材溶液各10μL,分别点于同一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以氯仿-甲醇-甲酸甲酯-水 (3︰4︰8︰2)为展开剂,展开,取出,晾干,喷以5%硫酸乙醇溶液,105℃加热5 min,置紫外灯(365 nm)下检视。供试品色谱中,在与金樱子对照药材色谱相应位置上,显相同颜色的蓝色荧光斑点。
3 原儿茶酸含量测定
3.1 仪器与试药
3.1.1 仪器:日本岛津液相色谱仪 (Lcsolution Lite);数控超声波清洗机 (昆山市超声仪器有限公司);BP211D电子天平 (SARTORIUSAG)。
3.1.2 试药:色谱纯乙腈;色谱纯甲醇;水为纯净水;其它试剂均为分析纯;原儿茶酸 (批号:110809-200903,中检所);壮腰健肾小蜜丸 (某公司生产,批号:110302、110303、110304)。
3.2 流动相考察[5]流动相分别考察了乙腈︰0.055 mol/L的磷酸二氢钾溶液︰磷酸溶液 (10︰90︰0.2),甲醇︰水 (20︰80)两种流动相的检测效果,结果:以乙腈︰0.055 mol/L的磷酸二氢钾溶液︰磷酸溶液(10︰90︰0.2)为流动相所得的色谱图保留时间、以及样品的分离度较为良好,因此以乙腈︰0.055 mol/L的磷酸二氢钾溶液︰磷酸溶液(10︰90︰0.2)为流动相进行含量测定。
3.3 提取方法选择 供试品提取过程选取了4种萃取溶剂分别考察:甲醇︰1%冰醋酸溶液 (10︰90);甲醇︰1%冰醋酸溶液 (70︰30);甲醇︰1%冰醋酸溶液 (10︰90);乙酸乙酯。可见以乙酸乙酯为萃取溶剂时,狗脊中的原儿茶酸峰面积明显增大,提取率最大,因此选择乙酸乙酯为提取溶剂。
3.4 色谱条件与系统适应性 色谱柱:用十八烷基硅烷键合硅胶为填充剂;柱温:室温;以乙腈︰0.055 mol/L的磷酸二氢钾溶液︰磷酸溶液(10︰90︰0.2)为流动相;流速:1.0 mL/min;检测波长:260 nm;理论板数按原儿茶酸峰计算应不低于4 000。
3.5 干扰性试验 阴性样品对供试品 (批号:110302)无干扰,说明本测定原儿茶酸的含量无干扰。
3.6 线性关系试验
3.6.1 对照品溶液制备:取原儿茶酸对照品适量,精密称定,加甲醇制成浓度分别为5.313、10.63、21.25、42.5、85.0μg/mL的溶液,分别进针。
3.6.2 标准曲线:在上述色谱条件下,记录色谱图,测定峰面积。以对照品浓度 (μg/mL)为横坐标,以峰面积值为纵坐标作图,得到:原儿茶酸回归方程为:Y=76 798X+26 271,r=0.999 9。
以上结果表明:在5.313μg/mL~85μg/mL范围内,原儿茶酸进样量与峰面积值线性关系良好,可用该方法测定原儿茶酸的含量。
3.7 日间精密度试验 按照配置相同浓度对照品溶液,进针,测定6次,根据峰面积计算RSD,RSD为1.0%,说明日间精密度良好。
3.8 供试品溶液制备
3.8.1 提取时间的考察:取本品5 g,剪碎,精密称定,置研钵中,加硅藻土2 g,研匀,转移至具塞锥形瓶中,用1%醋酸溶液10 mL分2次清洗研钵转移至具塞锥形瓶中,浸润,加乙酸乙酯50 mL,(1)超声10 min,残渣加乙酸乙酯30 mL超声10 min,过滤,合并续滤液;重复1次;(2)超声15 min,残渣加乙酸乙酯30 mL超声15 min,过滤,合并续滤液;重复1次;(3)超声20 min,残渣加乙酸乙酯30 mL超声20 min,过滤,合并续滤液;重复1次;之后各自将全部滤液置水浴中加热挥发干,残渣加甲醇适量使溶解,转移至5 mL量瓶中,加甲醇至刻度,摇匀,用微孔滤膜 (0.45μm)滤过,取续滤液,即得。测定含量,结果见表1。

表1 提取时间的比较
以上结果表明,提取时间为45 min,样品中原儿茶酸提取已经完全,故确定提取时间为45 min。
3.8.2 溶剂用量的考察:取本品5 g,剪碎,精密称定,置研钵中,加硅藻土2 g,研匀,转移至具塞锥形瓶中,用1%醋酸溶液10 mL分2次清洗研钵转移至具塞锥形瓶中,浸润。(1)加乙酸乙酯30 mL,超声15 min,滤过;(2)加乙酸乙酯50 mL,超声15 min,滤过;(3)加乙酸乙酯70 mL,超声15 min,滤过,上述3种残渣再加乙酸乙酯30 mL,超声处理15 min,过滤、合并滤液,重复1次,之后将全部滤液置水浴中加热蒸干,残渣加甲醇4 mL使溶解,转移至5 mL量瓶中,加甲醇至刻度,摇匀,用微孔滤膜 (0.45μm)滤过,取续滤液,即得。测定其含量,结果见表2。

表2 提取溶剂用量的比较
以上结果表明,第1次提取溶剂用量为50 mL,样品中原儿茶酸含量不再增加,因此选择提取溶剂量为50 mL。
3.9 重复性试验 采用我公司拟定含量测定方法,对样品 (批号:110302)进行处理,分别制备5份,各进样20μL,依据峰面积值进行计算,RSD为1.1%。
3.10 回收率试验 称取已知含量的样品 (批号:110302)约2.5 g,平行9份,其中3份分别加入浓度为0.027 mg/mL原儿茶酸对照品溶液各1 mL,3份分别加入0.047mg/mL原儿茶酸对照品溶液各1 mL,3份分别加入0.033 5 mg/mL的原儿茶酸对照品溶液各2 mL,照“供试品溶液制备”项下操作,测定,以下式计算方法得回收率。回收率(%)=(测得总量-样品含量)/加入量×100%。结果见表3。
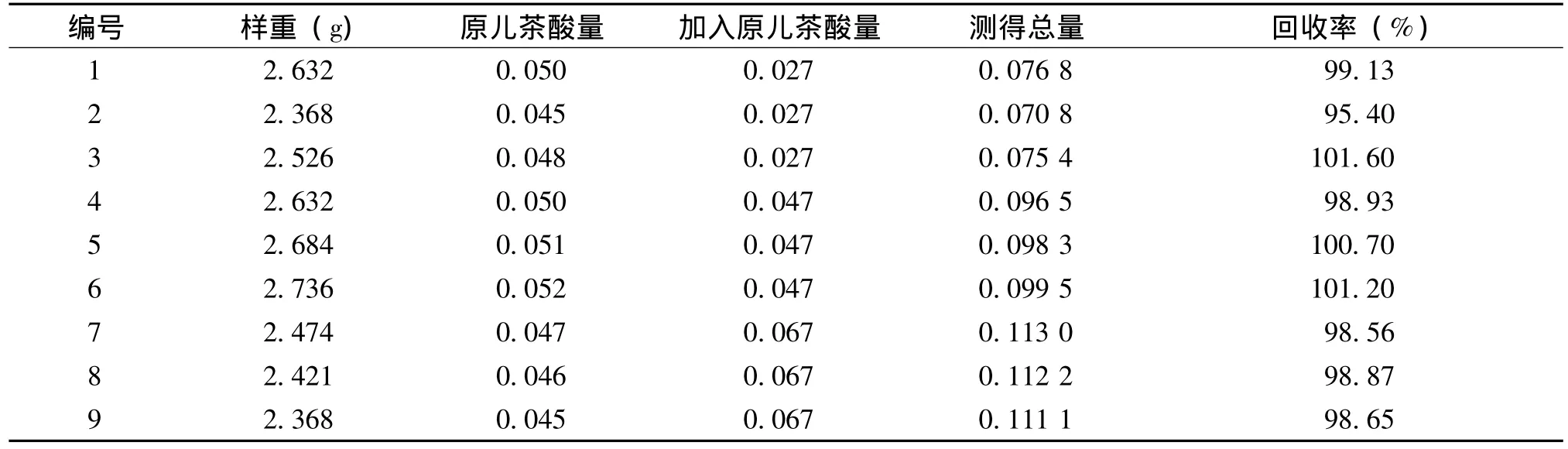
表3 回收率试验结果 (mg)
试验结果表明:平均回收率为99.22%,RSD为1.9%,加样回收率良好。
3.11 溶液稳定性试验 称取样品 (批号:110302)约5 g,精密称定,照某公司确定的方法制备溶液,取上述溶液,分别于0、2、6、12 h和24 h时,进针,测得样品中原儿茶酸峰面积值,RSD为1.2%,证明样品溶液在24 h内检测稳定。
3.12 十批样品中试样品检验结果 详见表4。

表4 十批样品检验结果
3.13 样品中原儿茶酸含量限度的确定 该品种是小蜜丸,采用狗脊提取物入药,考虑到药材来源不同,根据十批次成品检测结果,最低一次为16 μg/g,在此基础上下浮30%,故正文中暂订本品每1 g小蜜丸含狗脊以原儿茶酸 (C7H6O4)计不得少于11μg。
4 讨论
4.1 牛大力、黑老虎、鸡血藤供试品提取方法参考壮腰健肾片和舒筋活血片的质量标准下的方法,展开剂由苯-丙酮-甲酸 (5︰1︰0.2)改为甲苯-乙酸乙酯-甲酸 (10︰7︰1),减少毒性;三者一起薄层鉴别,在荧光下显现不同颜色斑点;方法简单独特,重现性良好,并且节省人力物力,值得推广。
4.2 色谱柱选择 采用所选流动相考察了不同厂家的两根色谱柱,原儿茶酸在甲柱 (origisil C18,5μm,250×4.6 mm)保留时间13 min左右,乙柱 (Promosil C18,5μm,250×4.6 mm)保留时间7 min左右,考虑到该样品成分较多,选用保留时间较长的色谱柱可以得到良好分离效果,因此选择甲柱进行试验。
4.3 原儿茶酸含量测定方法参照壮腰健肾片的流动相进行了预实验,发现分离度不好,可能是壮腰健肾小蜜丸与壮腰健肾片含有的辅料不同,影响到狗脊中原儿茶酸的分离。
[1] 张春辉,吴爱英,杨晓云,等.壮腰健肾片的质量标准[S].中国药师,2006,12(9):1 121-1 123
[2] 程世云,班永生.舒筋活血片质量标准研究[S].安徽医学,2011(5):38-40
[3] 李亚萍,庄义修,雷晓林.治带胶囊质控方法研究 [J].淮海医药,2007(4):85-86
[4] 原红霞,裴妙荣,杨素德.治带片的定性定量研究 [J].中国实验方剂学,2010(1):32-34
[5] 张善玉,姜艳玲,朴惠顺,等.HPLC测定老鹳草中的原儿茶酸[J].华西药学杂志,2007(4):92-93
(2014-12-07收稿)
猜你喜欢
现代食品科技(2022年5期)2022-05-30
湖北科技学院学报(医学版)(2021年5期)2021-12-29
昆明医科大学学报(2021年8期)2021-08-13
环境卫生工程(2021年3期)2021-07-21
环境卫生工程(2020年3期)2020-07-27
供水技术(2020年6期)2020-03-17
天然产物研究与开发(2018年8期)2018-09-10
中成药(2018年6期)2018-07-11
资源节约与环保(2018年1期)2018-02-08
中成药(2017年8期)2017-11-22
