
基于关键节点子团的乳腺癌候选疾病模块挖掘算法
2016-06-22 06:59王一斌程咏梅张绍武
东南大学学报(自然科学版) 2016年2期
关键词:乳腺癌
王一斌 程咏梅 张绍武
(西北工业大学信息融合技术教育部重点实验室, 西安 710072)
基于关键节点子团的乳腺癌候选疾病模块挖掘算法
王一斌 程咏梅 张绍武
(西北工业大学信息融合技术教育部重点实验室, 西安 710072)
摘要:为解决乳腺癌疾病模块挖掘方法中基因表达谱样本数量少、数据不完整、存在噪声和偏差的问题,提出了一种基于关键节点子团和局部适应度的候选疾病模块挖掘算法——KNGLF算法.该算法首先将候选基因与致病基因间的重叠相似性得分和功能相似性得分进行融合,通过比较融合得分与阈值,筛选出关键节点,并构建关键节点子团;然后,基于局部适应度及不同节点对应的不同判定标准,扩展挖掘候选疾病模块;最后,根据富集分析结果确定候选疾病基因模块.实验结果表明,与现有其他乳腺癌模块挖掘算法相比,KNGLF中关键节点选择算法所得平均排名较小,曲线下面积较大.KNGLF算法挖掘出15个具有较显著生物意义的乳腺癌候选疾病模块.此外,KNGLF算法还可扩展至其他疾病候选模块.
关键词:乳腺癌;疾病模块挖掘;候选基因打分;关键节点子团;局部适应度
癌症是一种细胞失控增长的复杂多发疾病,在全世界范围内已成为一个重要的公共健康问题[1].在各类型癌症中,乳腺癌是全球女性最常见的恶性肿瘤,世界上绝大多数国家在过去20年中发病率都持续增长[2].以统计生化试验的方法来寻求疾病分子靶点进行治疗,虽然取得了一定的成果,但大都以特定的基因目标为试验对象,因而所得结果有限,且需消耗大量时间和人力物力.
通过生物网络挖掘疾病功能模块,不仅能为分子靶点研究提供有效的信息,还能展示疾病基因及其产物以及它们彼此之间的协同关系,全面阐明其在复杂疾病过程中的作用机理.目前,学者们已提出了多种基于网络的乳腺癌疾病模块挖掘方法.Yang等[3]提出了一种基于路径的乳腺癌核心模块挖掘方法,利用该方法虽然辨识出了一些与该疾病有关的生物标记和功能模块,但由于主要依靠基因表达谱数据,故易受表达数据缺失、冗余和偏差以及样本数量有限的影响,且模块构建较为简单,没有进行更深入的筛选.Jia等[4]利用已有工具从构建出的局部共表达网络中挖掘模块,然后结合差异表达基因和显著样本的分布特性来筛选出癌症风险模块,通过与已知癌症样本之间的风险关系来判断模块的疾病风险程度;该方法仍局限于表达谱所涉及的基因,且使用统一的阈值来确定基因间连接关系,故而会导致一些弱相关性基因丢失.
鉴于此,本文提出了一种新的基于关键节点子团和局部适应度算法来挖掘乳腺癌候选疾病模块.该算法不使用基因表达谱数据,采用融合打分策略筛选出关键节点并构建关键节点子团,借助关键节点子团和局部适应度的思想进行模块挖掘,并根据富集分析来决定所挖掘模块是否为候选致病基因模块.
1材料和方法
1.1数据集
本文算法采用乳腺癌致病基因、人类蛋白质相互作用(PPI)、表型相似性及GO四个数据集.致病基因数据集由3部分组成,其中2部分来源于BCDB(BreastCancerGeneDataBase)数据库和G2SBC(TheGenes-to-SystemsBreastCancer)数据库,另一部分从OMIM(OnlineMendelianInheritanceinMan)GeneMap数据库筛选得到.人类蛋白质相互作用数据来源于HPRD(HumanProteinReferenceDatabase)数据库.表型相似性数据来自文献[5].
利用致病基因集和表型相似性数据集,构造候选基因集.首先,将一个致病基因相对应的表型MIM编号映射到表型相似性得分矩阵中,并将该表型与其他所有表型的相似性得分平均值作为阈值.然后,挑选相似性得分大于此阈值的其他表型作为候选表型,并在OMIM中找到候选表型所对应的基因.最后,对候选表型所涉及的每一个基因进行连锁间隔长度为10的关联查询,构建候选基因.对每一个乳腺癌致病基因进行同样的处理,构成候选基因集.
1.2KNGLF算法
1.2.1关键节点选择
对某个候选基因g,它与致病基因集V之间存在3个度量值:平均两节点拓扑重叠度量值Tag(V,g)、多节点拓扑重叠度量值Tmg(V,g)、GO注释间的功能相似性度量值sim(V,g).这3个值可融合成综合得分函数F(V,g),即
F(V,g)=αTag(V,g)+βTmg(V,g)+γsim(V,g)
(1)
式中,α,β,γ为加权系数.
令阈值为tc,若F(V,g)≥tc,则该候选基因g被视为关键节点.下面就Tag(V,g),Tmg(V,g),sim(V,g)和tc的定义及计算方法进行详细描述.
1) 平均两节点间拓扑重叠相似性度量
在无权网络中,节点i,j之间的拓扑重叠相似性度量定义为[6]
(2)

下面对邻居节点的范畴进行扩展.所用的邻居节点中加入了与源节点之间步长为二步和三步的节点,即与源节点之间通过1个或2个节点相连的节点.考虑到PPI网络的计算复杂度,设定最大步长为3,即分别计算一步、二步和三步邻居节点条件下候选基因g与乳腺癌致病基因集V之间的平均两节点拓扑重叠度量值at1(V,g),at2(V,g)和at3(V,g),计算公式如下:
(3)
式中,n为致病基因集V中致病基因数目;d为V中的一个致病基因;m为节点间步长;tm(d,g)为不同步长条件下d,g两节点间的拓扑重叠相似性度量.
将3种不同条件下的平均两节点拓扑重叠度量值进行归一化并加以融合,得到候选基因g与致病基因集V之间的平均两节点拓扑重叠度量值
Tag(V,g)为
Tag(V,g)=xat1(V,g)+yat2(V,g)+zat3(V,g)
(4)
式中,x,y,z为融合系数.
2) 多节点间拓扑重叠相似性度量
对于某个候选基因g和致病基因集V,一步、二步和三步邻居节点条件下的多节点拓扑重叠度量值mt1(V,g),mt2(V,g)和mt3(V,g)的计算公式为
mtm(V,g)=tm(d1,d2,…,dn-1,dn,g)=
(5)
式中,{d1,d2,…,dn}∈V.
将3种不同条件下的多节点拓扑重叠度量值归一化并融合,得到候选基因g与整体致病基因集V之间的多节点拓扑重叠度量值Tmg(V,g)为
Tmg(V,g)=xmt1(V,g)+ymt2(V,g)+zmt3(V,g)
(6)
3) 基因之间功能相似性度量
一个基因可用一个或多个GO术语进行描述.对于某个GO术语a而言,其深度信息为
DP(a)=SP(a,R)
(7)
式中,SP为2个GO术语间的最短路径,表示在有向无环图中连接2个节点所经过的最少边数;R表示根目录,即GO术语中3个最基本根目录(分子功能、生物过程和细胞成分)中的任意一个.
对于任意给定的2个GO术语a和b,若c为a和b公共的父节点,则a和b间的功能相似度度量值为
(8)
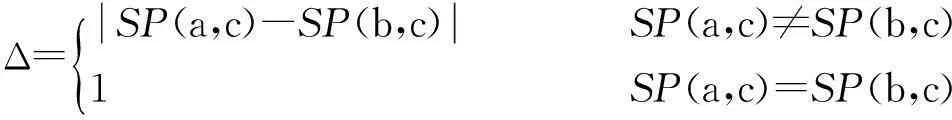
Δ=SP(a,c)-SP(b,c) SP(a,c)≠SP(b,c)1 SP(a,c)=SP(b,c){
式中,f1,f2分别为DP,SP对功能相似度贡献率的调节参数,且f1,f2∈(0,1).
一个基因J可用多个术语进行注释.单个术语和单个基因的功能相似性度量值定义为
(9)
式中,Tl∈J={T1,T2,…,Tp};To为任一术语且To∉J.
假设2个基因JA和JB,令{TA1,TA2,…,TAh}和{TB1,TB2,…,TBq}分别为其所对应的术语集,JZ为JA和JB的交集,JA-JZ为JA和JZ的差集,JB-JZ为JB和JZ的差集,则JA和JB之间的功能相似性度量值为
(10)
式中,Tz为JZ中的术语;Ts为JA和JZ差集中的术语;Tt为JB和JZ差集中的术语;h,q分别为基因JA和JB所包含的注释术语数目.
基因间功能相似性取值范围为[0,1],GO术语与自身的功能相似性度量值定义为
(11)
由此便可计算候选基因g和乳腺癌致病基因集V间的功能相似性度量值为
(12)
1.2.2模块和节点局部适应度
对于一个有权或无权网络G(V,E),令其中某一模块H中节点v(H⊆G)的加权入度为din(H,v),表示模块H中所有与节点v直接相连的节点的边权重总和;令节点v的加权出度为dout(H,v),表示属于G但不属于模块H的节点与节点v直接相连的边权重总和,即
式中,u为网络中的节点;w(u,v)表示与v直接相连的边权重.
模块H的局部适应度fH为
(13)
对于模块H中的任一节点v,其节点局部适应度fHv定义为节点v加入模块H前后模块H的局部适应度变化之差,即
fHv=fH∪{v}-fH
(14)
式中,fHU{v},fH分别表示模块H包含、不包含节点v时的局部适应度.
1.2.3算法步骤
KNGLF算法的详细步骤如下:
① 针对PPI网络,构建独立的关键节点子团集合{GK1,GK2,…,GKi,…,GKm},且各关键节点子团中要求包含2个及2个以上节点.
② 在PPI网络中,寻找关键节点子团集合中某一关键节点子团GKi的邻居节点集合M.
③ 对M中不同节点采用不同的判断标准.若M中包含有GKi以外的其他关键节点集合{K1,K2,…,Ki,…,Kn},则转至步骤④;否则,转至步骤⑤.
④ 计算关键节点集合中某个关键节点Ki对子团GKi的节点局部适应度fGki.若fGki>0,则将该节点添加到关键节点子团GKi中,更新关键节点子团并返回步骤②;否则,舍弃该节点,从关键节点集合选择剩余的其他关键节点,继续比较节点局部适应度,直至遍历集合{K1,K2,…,Ki,…,Kn}中所有关键节点.
⑤ 计算M中所有非关键节点对关键节点子团GKi的节点局部适应度,令节点局部适应度最大的非关键节点为P′,如果fGKi>tm(tm为所设阈值),则将该节点添加到子团GKi中,更新子团并返回步骤②;否则,子团GKi停止扩张,该模块挖掘结束,选择关键节点子团集合中下一个子团并返回步骤②进行模块挖掘工作,直至遍历集合{GK1,GK2,…,GKi,…,GKm}中的每一个子团.
1.2.4参数选取
对于已知包含n个致病基因的乳腺癌致病基因集,预设了一系列融合系数,并采用留一法进行验证,得到不同参数情形下各致病基因排名,选取各情形下排名前20%的基因,并从中统计致病基因数目,选取数目最多时所对应的参数为最优融合参数.对于式(1),选取α=3,β=1,γ=4;对于式(4)和 (6),选取x=5,y=2,z=1;对于式(8)选取f1=0.8,f2=0.1.
利用留一法进行验证的过程中,将各种参数条件下的致病基因进行排名,分别计算不同条件下排名前90%的各致病基因的综合得分,并将得分平均值作为关键节点的选取阈值tc.利用PPI网络中现有的连接关系,将乳腺癌已知致病基因构建成若干个规模不同的模块,分别计算这些模块的适应度,选取平均模块适应度值作为非关键节点能否加入模块的阈值tm.
2结果和讨论
2.1挖掘结果及部分富集分析
在致病基因集中,从BCGB,G2SBC和OMIM数据库中分别收集了62,44和48个致病基因,剔除重复数据后,获得138个乳腺癌致病基因.利用表型间相似性构建相应候选基因集,通过打分筛选,最终得到1 935个关键节点,并构建出各关键节点子团.采用KNGLF算法挖掘候选疾病模块,应用在线工具Go-TermFinder进行富集分析和确认,显著性阈值P默认为0.01,最终获得15个具有一定生物学意义乳腺癌候选疾病模块.
以排名第1的关键节点子团所挖掘出的TZMMZ候选疾病模块为例,利用生物信息数据平台DAVID[7],分别在生物过程、细胞成分和分子功能以及KEGG通路中进行富集条目(P<0.01)分析.从分析结果中发现,该模块的富集条目主要集中在调控过程,如血管再生、某些复合物的合成过程、新陈代谢过程和细胞活动方面以及部分转录化合物的活性和绑定功能方面.其中,GO:0045766和GO:0001525涉及到血管再生,尤其是对乳腺组织的血管再生和增殖;GO:0061180涉及乳腺上皮细胞发展和乳腺增生过程,文献[8]也提出该注释相关的血管内皮增长因子与乳腺极性组织的缺失有关.在KEGG通路富集分析中,hsa04110直接磷酸化和激活了肿瘤抑制蛋白p53,而p53与其转录目标在乳腺癌中其重要作用;ko03440同源重组为DNA双链损伤精确修复的关键,与乳腺癌相关的抑癌基因通过同源重组共同起作用.由此可以推测,该模块可能对乳腺癌抑制细胞起负作用,同时激活癌细胞并使其失控增长.
2.2算法比较
对于乳腺癌致病基因集,分别采用KNGLF中关键节点选择算法、重启随机游走 (randomwalkwithrestart,RWR)算法[9]、Wu等[10]提出的CIPHER算法以及关键节点选择算法中单一的相似性度量算法(平均两节点拓相扑重叠相似度量算法、多节点拓扑重叠相似度算法和基因间功能相似度量算法)对致病基因平均排名比[11](meanrankratio,MRR)进行计算和比较,结果见表1.由表可知,KNGLF中关键节点选择算法的平均排名比最小,优于关键节点选择算法中单一的相似性度量算法、RWR算法和CIPHER算法.图1为前3种算法的ROC曲线.由图可知,KNGLF中关键节点选择算法、CIPHER算法和RWR算法的曲线下面积(AUC)分别为93.37%,88.27%和84.24%.

表1 乳腺癌致病基因在不同打分算法下的平均排名比

图13种算法的ROC曲线


表2 基于3种算法的乳腺癌模块挖掘结果
文献[4]利用乳腺癌基因表达谱数据GSE20437和所挖掘出的乳腺癌风险模块,依据所提的样本疾病风险评估算法对每个样本进行评估打分,并按得分高低进行排序,从而得到不同情形下的真阳性率(truepositiverate,TPR)、假阳性率(falsepositiverate,TPR)及其相应的ROC曲线.使用相同的乳腺癌表达谱数据和评估方法,结合KNGLF算法所得的乳腺癌候选疾病模块绘制出相应的ROC曲线(见图2).由图可知,文献[4]算法和KNGLF算法的乳腺癌样本风险评估ROC曲线下面积分别为89.29%和78.62%.相比于文献[4]算法所得的乳腺癌样本风险评估ROC曲线面积,KNGLF算法高出10.67%.因此,KNGLF算法明显提高了乳腺癌候选疾病模块挖掘的准确性和可靠性.
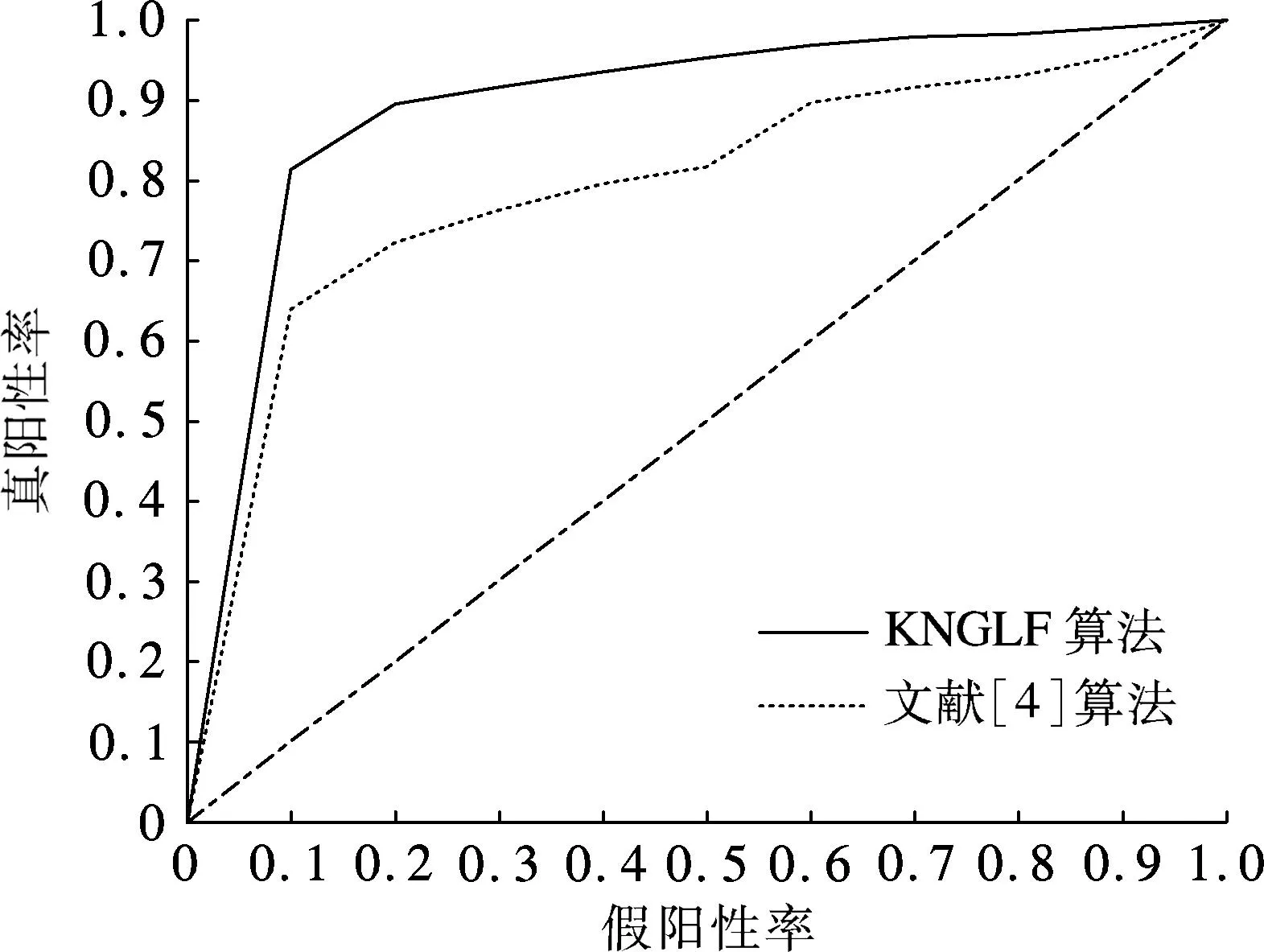
图2 乳腺癌基因表达谱评估ROC曲线
3结语
针对利用基因表达谱数据进行乳腺癌疾病模块挖掘时易受样本数量少、数据不完整影响、存在噪声和偏差的问题,提出了一种基于关键节点子团和局部适应度的模块挖掘KNGLF算法.该算法未采用基因表达谱数据,而是通过整合疾病表型信息、GO深度信息、PPI网络等对候选基因进行打分,筛选关键节点来构建关键节点子团,使用局部适应度和富集显著性分析思想进行模块挖掘和确认.实验结果表明,利用KNGLF算法所挖掘出的模块不仅有较好的富集显著性,且与乳腺癌疾病密切相关,在该疾病的发生和发展过程中起到了一定的作用和影响.相比于其他打分策略,KNGLF算法打分策略得到的致病基因平均排名较低,AUC值大于RWR算法和CIPHER算法;与其他模块挖掘算法相比,KNGLF算法挖掘的模块不仅包含较多核心基因数目,而且在模块数目、平均模块P值、模块Top5平均P值、模块Top10平均P值、平均模块密度等方面具有明显优势.由此可知,KNGLF算法不依赖于表达谱数据,可准确挖掘出具有一定生物意义的乳腺癌候选模块,对疾病病理了解、前期诊断、后期预防和治疗提供了较大帮助,还可进一步扩展用于挖掘其他疾病候选模块.
参考文献 (References)
[1]KimSY,HahnWC.Cancergenomics:Integratingformandfunction[J]. Carcinogenesis, 2007, 28(7): 1387-1392.DOI:10.1093/carcin/bgm086.
[2]EarlyBreastCancerTrialists’CollaborativeGroup.Adjuvantbisphosphonatetreatmentinearlybreastcancer:Meta-analysesofindividualpatientdatafromrandomisedtrials[J]. The Lancet, 2015, 386(10001): 1353-1361.DOI:10.1016/S0140-6736(15)60908-4.
[3]YangR,DaigleBJJr,PetzoldLR,etal.Coremodulebiomarkeridentificationwithnetworkexplorationforbreastcancermetastasis[J]. BMC Bioinformatics, 2012, 13(1): 12-1-12-11.DOI:10.1186/1471-2105-13-12.
[4]JiaX,MiaoZ,LiW,etal.Cancer-riskmoduleidentificationandmodule-baseddiseaseriskevaluation:Acasestudyonlungcancer[J]. PloS ONE, 2014, 9(3):e92395.DOI:10.1371/journal.pone.0092395.
[5]vanDrielMA,BruggemanJ,VriendG,etal.Atext-mininganalysisofthehumanphenome[J]. European Journal of Human Genetics, 2006, 14(5): 535-542.DOI:10.1038/sj.ejhg.5201585.
[6]LiA,HorvathS.Networkneighborhoodanalysiswiththemulti-nodetopologicaloverlapmeasure[J]. Bioinformatics, 2007, 23(2): 222-231.DOI:10.1093/bioinformatics/btl581.
[7]AlvordG,RoayaeiJ,StephensR,etal.TheDAVIDgenefunctionalclassificationtool:Anovelbiologicalmodule-centricalgorithmtofunctionallyanalyzelargegenelists[J]. Genome Biol, 2007, 8(9):R183.DOI:10.1186/gb-2007-8-9-r183.
[8]ChenA,CuevasI,KennyPA,etal.Endothelialcellmigrationandvascularendothelialgrowthfactorexpressionaretheresultoflossofbreasttissuepolarity[J]. Cancer Rresearch, 2009, 69(16): 6721-6729.DOI:10.1158/0008-5472.CAN-08-4069.
[9]KöhlerS,BauerS,HornD,etal.Walkingtheinteractomeforprioritizationofcandidatediseasegenes[J]. The American Journal of Human Genetics, 2008, 82(4): 949-958.DOI:10.1016/j.ajhg.2008.02.013.
[10]WuX,JiangR,ZhangMQ,etal.Network-basedglobalinferenceofhumandiseasegenes[J]. Molecular Systems Biology, 2008, 4(1): 189-1-189-11.DOI:10.1038/msb.2008.27.
[11]JiangR,GanM,HeP.Constructingagenesemanticsimilaritynetworkfortheinferenceofdiseasegenes[J]. BMC System Biology, 2011, 5(S2):S2-1-S2-11.DOI:10.1186/1752-0509-5-S2-S2.
Miningalgorithmforbreastcancercandidatediseasemodulebasedonkeynodegroups
WangYibinChengYongmeiZhangShaowu
(KeyLaboratoryofInformationFusionTechnologyofMinistryofEducation,NorthwesternPolytechnicalUniversity,Xi’an710072,China)
Abstract:In order to solve the problems of small quantity, incomplete data, noise, and bias of the gene expression profile in the method for breast cancer disease module mining, a mining algorithm for candidate disease module based on the key node groups and the local node fitness constraints, the key node groups and local fitness (KNGLF) algorithm, is proposed. First, the topological overlap similarity score and the functional similarity score between the candidate genes and the pathogenic genes are fused into a fusion score. Through comparing the fusion score with the threshold value, the key nodes are selected and the key node groups are constructed. Then, the breast cancer candidate disease modules are mined based on the local fitness constraints and different decision criteria for different nodes. Finally, according to the enrichment analysis results, the candidate disease gene modules are identified. The experimental results show that compared with other existing mining algorithms for breast cancer module, the key node selection algorithm in the KNGLF algorithm has the smaller MRR (mean rank ratio) but the greater AUC (area under curve). Fifteen breast cancer candidate gene modules with significant biological significance are identified by the KNGLF algorithm. Besides, the KNGLF algorithm can be extended to identify other diseases related candidate modules.
Key words:breast cancer; disease module mining; candidate gene score; key node groups; local fitness
DOI:10.3969/j.issn.1001-0505.2016.02.007
收稿日期:2015-07-15.
作者简介:王一斌(1982—),男,博士生;张绍武(联系人),男,博士,教授,博士生导师,zhangsw@nwpu.edu.cn.
基金项目:国家自然科学基金资助项目(61170134,61473232,91430111)、国家自然科学基金青年基金资助项目(61502396)、互联网金融创新及监管四川省协同创新中心资助项目.
中图分类号:R318.04;Q78
文献标志码:A
文章编号:1001-0505(2016)02-0265-06
引用本文: 王一斌,程咏梅,张绍武.基于关键节点子团的乳腺癌候选疾病模块挖掘算法[J].东南大学学报(自然科学版),2016,46(2):265-270.DOI:10.3969/j.issn.1001-0505.2016.02.007.
猜你喜欢
中老年保健(2022年6期)2022-08-19
现代临床医学(2022年1期)2022-02-12
中老年保健(2021年3期)2021-08-22
中国生殖健康(2019年2期)2019-08-23
中国生殖健康(2019年6期)2019-01-06
中国生殖健康(2019年5期)2019-01-06
幸福家庭(2019年14期)2019-01-06
祝您健康(2018年5期)2018-05-16
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
中国卫生标准管理(2015年6期)2016-01-14
