
分子动力学模拟研究直链淀粉与α-亚油酸的包合行为
2016-12-26 06:21庄海宁FengChenBruceHamakerOsvaldoCampanella
中国粮油学报 2016年4期
冯 涛 王 旭 庄海宁 Feng Chen Bruce R Hamaker Osvaldo Campanella 王 凯
(上海应用技术学院香料香精技术与工程学院1,上海 201418)(上海市农业科学院食用菌研究所 国家食用菌工程技术研究中心2,上海 201403)(美国普度大学食品系惠斯特勒碳水化合物研究中心3,美国印第安纳州 47906-2009)(云南中烟工业有限责任公司技术中心4,昆明 650231)
分子动力学模拟研究直链淀粉与α-亚油酸的包合行为
冯 涛1王 旭1庄海宁2Feng Chen3Bruce R Hamaker3Osvaldo Campanella3王 凯4
(上海应用技术学院香料香精技术与工程学院1,上海 201418)(上海市农业科学院食用菌研究所 国家食用菌工程技术研究中心2,上海 201403)(美国普度大学食品系惠斯特勒碳水化合物研究中心3,美国印第安纳州 47906-2009)(云南中烟工业有限责任公司技术中心4,昆明 650231)
α-亚油酸和直链淀粉可以形成热力学稳定复合物。为了研究α-亚油酸影响直链淀粉构象的机理、复合物的构象趋势及复合作用过程中氢键的变化情况,在373 K条件下进行了长时间(500 ns)的全原子分子动力学模拟。模拟发现在有/无α-亚油酸存在的情况下,在500 ns内都可以观察到大量有序到无序的构象转变,这表明直链淀粉和α-亚油酸之间结合作用微弱。原子均方根偏差(RMSD)、α-亚油酸与直链淀粉轴心间距、氢键增加都证明直链淀粉-α-亚油酸复合物的形成是热力学自发行为。
分子动力学模拟 直链淀粉 α-亚油酸
直链淀粉和脂类可以形成复合物。复合配体物的存在可以导致直链淀粉形成结构紧密的螺旋结构。该螺旋具有一个疏水性的空腔。一般认为脂类分子的非极性端进入螺旋空腔,羧基端则在螺旋外部[1]。螺旋结构的形成可能与配体物在螺旋空腔内具有更低的自由能有关。该驱动力应该是与直链淀粉尽量减少其与水的接触面积有关[2]。分子内键合力,例如范德华力和氢键主要存在于螺旋上的螺圈之间,以稳定单一螺旋链[3-4]。分子间作用力则稳定直链淀粉与其配体之间的相互作用[5]。
直链淀粉-脂肪酸复合物的结晶态是V-直链淀粉6倍(即6个葡萄糖残基)单链左手螺旋[6]。Zabar等[7]从分子、纳米和微米尺度研究了直链淀粉-长链脂肪酸复合物,但未达到原子水平。虽然大量的试验数据证明了直链淀粉与α-亚油酸可以形成很稳定的复合物,但仍然有一些问题需要在原子水平上进行进一步研究,这就需要借助分子动力学模拟来实现。分子动力学模拟属于分子模拟的一种,随着计算机技术的快速发展,其已逐渐成为预测体系稳定性、验证理论假设及改进模型的重要工具。目前分子动力学模拟在直链淀粉研究方面的应用已有不少成果[1, 8-9]。
本研究拟在有/无α-亚油酸的条件下,利用分子动力学对α-亚油酸与直链淀粉的复合过程进行动态模拟,以从原子水平解释以下问题:游离脂肪酸稳定直链淀粉构象的原因;直链淀粉-α-亚油酸复合物的热稳定构象状态;在复合物形成过程中氢键的变化。
1 材料与方法
1.1 直链淀粉和α-亚油酸模型的选取
V-直链淀粉的分子模型由Paderborn大学提供。该直链淀粉模型为55个葡萄糖残基组成的6倍左手螺旋。所得的螺旋其内径为54 nm,外径为135 nm,长738 nm。根据糖苷键链接不同,GLC、GLM和GLK分别用于代表直链淀粉的起始、中间和末端葡萄糖残基。α-亚油酸的PDB文件从http://xray.bmc.uu.se/hicup/EIC/下载。具有20个原子的PDB文件选自MSDchem数据库。α-亚油酸的起始结构由PRODRG服务器生成(http://davapc1.bioch.dundee.ac.uk/prodrg/)。EIC表示α-亚油酸。
1.2 分子力场
葡萄糖分子力场根据Damm等[10]和Kony等[11]改写。所有的模拟均根据为直链淀粉设计的葡萄糖分子力场来进行运算。原子上的电荷如表1所示。
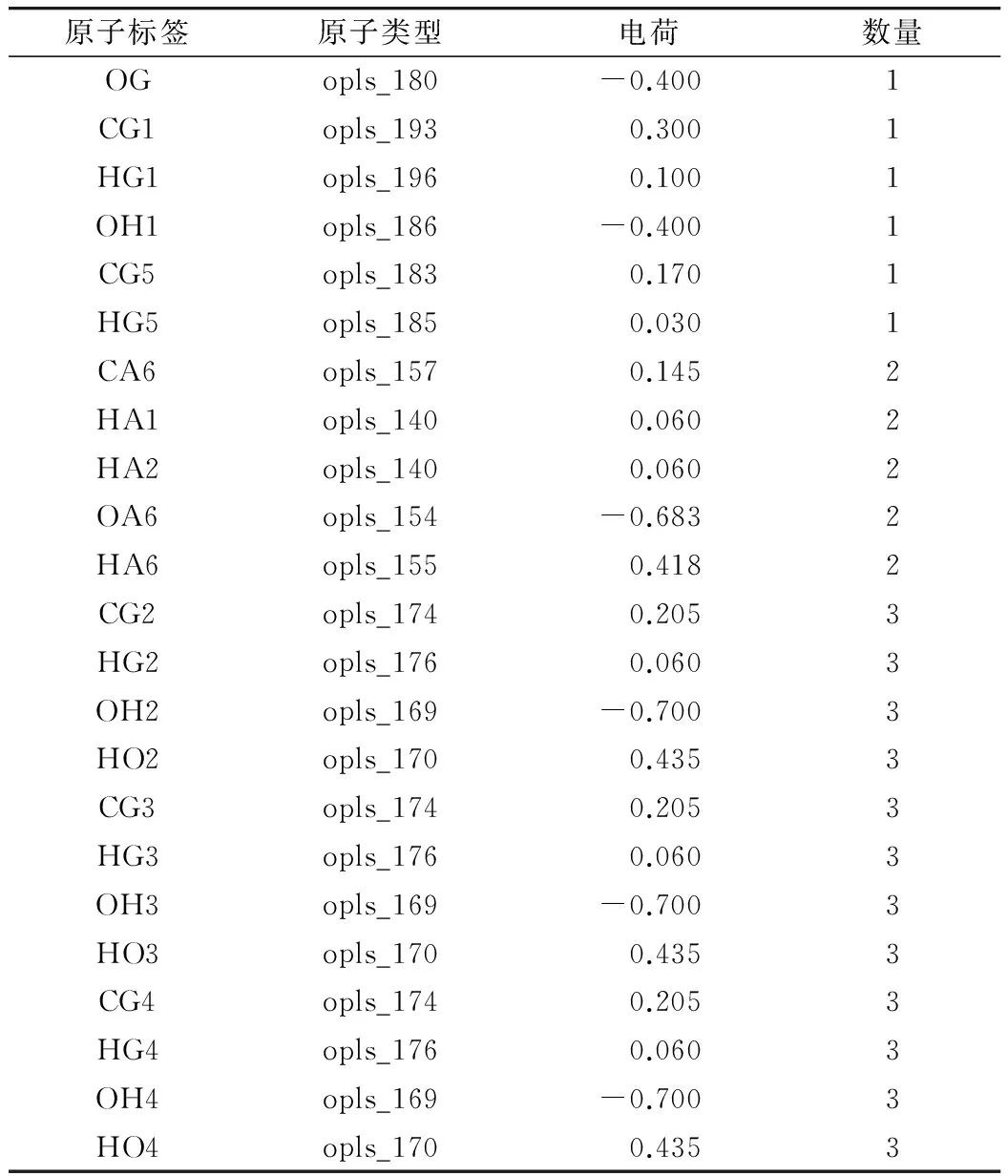
表1 葡萄糖拓扑结构中原子的电荷情况
注:OG,CG1~CG5,HG1~HG5分别代表葡萄糖环状结构上的氧,碳和氢原子;OH1~OH4,代表葡萄糖环状结构上的羟基氧原子;HO2~HO4,代表葡萄糖环状结构上的羟基氢原子;CA6,代表葡萄糖环状结构上的6位碳原子;OA6,代表葡萄糖环状结构上的6位氧原子;HA1,HA2,代表与葡萄糖环状结构上的6位碳原子相连的两个氢原子;HA6,代表与葡萄糖环状结构上的6位氧原子相连的氢原子。与附图1中原子标签相对应。
1.3 分子模拟法则
使用Gromacs 4.6.1(ScalaLife Competence Center, the European Research Council)中的分子动力学软件包中的跳蛙移动算法,以2 fs作为步长。模拟步数为2 500 000 000步,因此总的模拟时间为500 ns。LINCS算法用于对所有的键长(特别是重原子-H键)进行条件约束,参数分别为lincs_iter=1,和lincs_order=4。温度通过修改的Berendsen恒热耦合在不同的基团之间以松弛时间为0.1 ps来维持在373 K。压力通过Parrinello-Rahman耦合压力浴在各向同性坐标下以松弛时间为2 ps来保持在1 bar。非键合作用采用临近网格切割图法处理。短程临近切割距离为0.9 nm,根据配对表每个步长都要进行1次评估。短程静电力切割半径为0.9 nm,长程范德华力切割半径为1.4 nm,亦根据每个步长的更新配对表分别自动进行评估[1]。
1.4 直链淀粉和直链淀粉复合物模拟与分析
试验进行了2种模拟,2组分子模拟都是从圆柱形的3D结构开始进行(葡萄糖形成的柱形3D结构的每个螺圈由6个葡萄糖组成)。第1种是将55个残基的直链淀粉链置于1个设计有12 631个水分子的矩形盒子里(10 nm×5 nm×5 nm)。水通过简单的TIP4P模型进行模拟[1]。2个模拟的起始构象如图1所示。在分子模拟开始前,链间能量采用最陡下降法最小化。然后模拟运行500 ns,旨在获得直链淀粉在水中的稳定构象。
第2种是由55个残基的直链淀粉与α-亚油酸形成的复合物系统。直链淀粉事先与α-亚油酸预先复合在一起。根据试验条件下的pH值,α-亚油酸的羧基端上的氢质子将完全解离,这使α-亚油酸带1个负电荷,加入1个Na离子以平衡这一负电荷。水通过简单的TIP4P模型进行模拟。直链淀粉起始采用V-直链淀粉构象,α-亚油酸的极性末端置于螺旋空腔内部。系统置于一个设计有12 631个水分子的矩形盒子里(10 nm×5 nm×5 nm)。模拟运行500 ns。
所有的模拟运算和分析均采用Gromacs 4.6.1来进行。
1.5 构象簇分析
为了从模拟的轨迹中找到结构中具有代表性的构象簇,本研究采用Daura等[12]描述的方法来进行。采用0.2 nm切割了直链淀粉相邻原子位置的RMSD值作为聚类中心点。该过程一直重复直到没有相关的轨迹被提取出来。所有计算由Gromacs 4.6.1来完成。
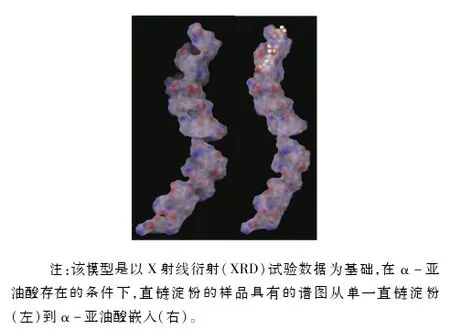
图1 由α-亚油酸诱导的直链淀粉的构象的可能模型
2 结果与讨论
2.1 直链淀粉复合物与单一直链淀粉之间的动态结构变化
图2(a,b)显示了每个体系(直链淀粉复合物和直链淀粉)在500 ns内的演变过程。2组模拟都展示了对于全结构的大量重新组合。在图2a中,在100 ns以内α-亚油酸首先从直链淀粉中分离出来,然后在200 ns时,α-亚油酸被重新包合进直链淀粉片段的中间区域,然后α-亚油酸始终被包合在直链淀粉片段内,但在200~500 ns的时间内,其位置在左右之间摇摆。直链淀粉则始终保持了一种伸展的左手螺旋的构象。但是,直链淀粉产生了一个完全折叠的构象,而且在500 ns的时间内始终扭曲缠绕,并且可以看出直链淀粉螺旋从片段的中点位置对折,且折叠构象不断变化。这说明直链淀粉上的原子移动得比直链淀粉复合物中的原子要剧烈得多。
淀粉骨架GLK_GLM_GLC&CA6与GLK之间的位移距离如图2c所示。在500 ns内,复合物与单独直链淀粉的平均模拟位移距离分别为1.0 nm和3.0 nm。这些位移距离轨迹表明在α-亚油酸的存在下,直链淀粉骨架上的原子移动得要缓慢些。α-亚油酸复合后的淀粉可以在200~500 ns内,维持一个相对稳定的构象,而单独直链淀粉在500 ns内为一个扭曲的相对不稳定的构象。因此,可以得出初步结论:单独的直链淀粉在水溶液中的构象是不稳定的,但在α-亚油酸的存在下,则可以极大地提高其稳定性。模拟的运动轨迹和试验现象(直链淀粉与α-亚油酸可以形成稳定络合物)的一致性说明分子动力学模拟为直链淀粉的构象转变提供了其受α-亚油酸影响的精确效应图。
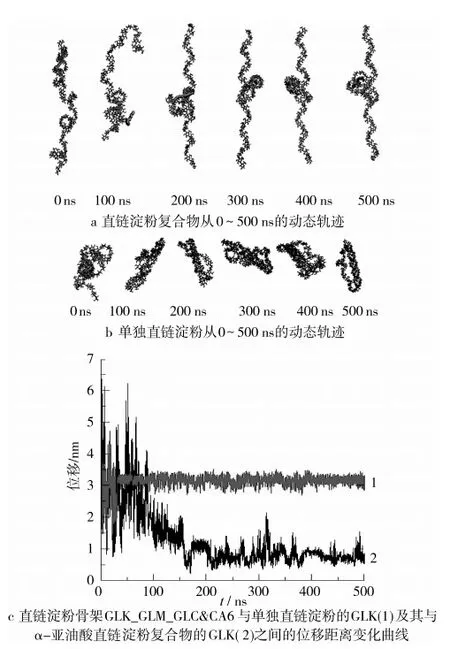
图2 有/无α-亚油酸条件下,直链淀粉的构象变化及其对应的淀粉骨架位移
2.2 直链淀粉复合物与单独直链淀粉的单一结构域的动态位移变化
直链淀粉骨架相对于GLC、GLK、GLM结构域位移的均方根偏差经计算如图3所示。可以看出,直链淀粉-α-亚油酸复合物的所有均方根偏差都完全小于单独直链淀粉,这意味着在直链淀粉-α-亚油酸复合物中的原子的动态位移变化要少于单独直链淀粉。2个体系中,骨架与GLM之间的RMSD最为光滑平坦,表明GLM结构域在3个结构域中是较为稳定的。反之,GLC和GLK均表现出了较大幅度的位移变化。其中,GLC的动态位移变化RMSD值在2个体系中均为最大,在复合物系统中为7.0 nm,在单独直链淀粉系统中为6.0 nm。但是,这一变化仅发生在前100 ns内,直链淀粉复合物的原子位移远比单独直链淀粉的剧烈。然后,当2个体系均达到平衡状态时,复合物系统的RMSD值均低于单独直链淀粉的。在2个体系中,GLK结构域的RMSD曲线遵循在图2c中所观察到的GLK-骨架之间的原子位移距离的变化趋势,在有α-亚油酸的条件下,其构象重排远比没有α-亚油酸的条件下温和。这些均表明α-亚油酸对直链淀粉的构象具有稳定作用。这些结果与图2中位移距离和动态轨迹的分析一致。

图3 直链淀粉骨架相对于GLC、GLK、GLM结构域位移的均方根偏差

图4 单独直链淀粉和复合直链淀粉的氢键数量
2.3 复合直链淀粉和单独直链淀粉的氢键数量
通常将受体-供体的距离低于0.35 nm和角度低于30°作为判断氢键形成的几何标准[13]。复合直链淀粉和单独直链淀粉与水分子之间形成的氢键数量的平均值分别为50和40(如图4)。这一结果说明复合直链淀粉在氢键的参与下聚合成紧凑的结构。氢键在聚合和维持复合直链淀粉的稳定结构中起很大作用。
2.4 复合直链淀粉和单独直链淀粉的主成分分析(PCA)
通过PCA分析来了解所观察到结构的主要累积波动图。图5表明了模拟的结构图形投影到平面上,该平面是由2个主要成分的共变量矩阵向量值所构建。在单独直链淀粉和复合直链淀粉模型中,所有沿轨迹方向的模拟系统均具有重叠的和分开的基本空间。在单独直链淀粉中,较宽的点集来自于GLM,在MD模拟的过程中,具有较为伸展和分散的区域大小。复合直链淀粉则具有一个较局限的、封闭的区域,表明构象变化幅度很小。在2个系统中,模拟的结构足以比较,覆盖了较宽泛的空间区域。单独直链淀粉通过具有较大的基本空间再次表明其最高的变化幅度。另外,为了进一步揭示在模拟期间,相应的残基之间的主要运动轨迹的幅度,单独直链淀粉作为PCA的参考模拟对象,并用其主成分投影所有的分子模拟轨迹。从单独直链淀粉的模拟体系中所得到的更为明显的现象支持了本研究的假设即α-亚油酸具有的稳定效应。

图5 2个模拟体系的主成份分析图
2.5 复合直链淀粉和单独直链淀粉最常见的构象簇
复合直链淀粉和单独直链淀粉的主要构象簇完全不同。图6显示了本模拟中复合直链淀粉和单独直链淀粉的典型构象簇图像。复合直链淀粉中较多的螺旋骨架基团导致了疏水性溶剂接触表面积的增加。这些构象簇也具有不同的水合半径值。最为伸展的构象(复合直链淀粉)比稍微不伸展的构象(单独直链淀粉)的水合半径要大50%。同样的结果,在三丙氨酸的卤盐溶液中的构象变化也被报道过[14]。

图6 2种模拟体系中最为常见的构象簇
3 结论
本研究在有/无α-亚油酸存在的条件下,模拟了直链淀粉的构象变化。模拟过程中α-亚油酸可以使直链淀粉处于一个比较稳定的构象,而单独直链淀粉的构象则是不断变化的。此外,在有/无α-亚油酸存在时,淀粉骨架相对于末端葡萄糖残基的位移变化、淀粉骨架单一结构域位移变化以及PCA结果都表明α-亚油酸可以稳定直链淀粉的构象。而氢键在聚合及维持直链淀粉-α-亚油复合物的结构中发挥很大作用。最后,本研究的结果也表明实时动力学在热力学稳定构型复合物的形成过程中的重要作用,同时对在原子水平上理解复合物行为具有重要意义。
[1]López C A, de Vries A H, Marrink S J. Amylose folding under the influence of lipids[J]. Carbohydrate Research, 2012, 364: 1-7
[2]Heinemann C, Conde-Petit B, Nuessli J, et al. Evidence of starch inclusion complexation with lactones[J]. Journal of Agricultural and Food Chemistry, 2001, 49(3): 1370-1376
[3]Banks W, Greenwood C. Amylose: a non-helical biopolymer in aqueous solution[J]. Polymer, 1971, 12(2): 141-145
[4]Karkalas J, Ma S, Morrison W R, et al. Some factors determining the thermal properties of amylose inclusion complexes with fatty acids[J]. Carbohydrate Research, 1995, 268(2): 233-247
[5]Lauro M, Poutanen K, Forssell P. Effect of partial gelatinization and lipid addition on α-amylolysis of barley starch granules[J]. Cereal Chemistry, 2000, 77(5): 595-601
[6]Buléon A, Delage M, Brisson J, et al. Single crystals of V amylose complexed with isopropanol and acetone[J]. International Journal of Biological Macromolecules, 1990, 12(1): 25-33
[7]Zabar S, Lesmes U, Katz I, et al. Studying different dimensions of amylose-long chain fatty acid complexes: Molecular, nano and micro level characteristics[J]. Food Hydrocolloids, 2009, 23(7): 1918-1925
[8]Tusch M, Krüger J, Fels G. Structural Stability of V-Amylose Helices in Water-DMSO Mixtures Analyzed by Molecular Dynamics[J]. Journal of Chemical Theory and Computation, 2011, 7(9): 2919-2928
[9]曾鲁红.分子动力学模拟研究直链淀粉的增塑[D]. 2012, 镇江: 江苏科技大学
[10]Damm W, Frontera A, Tirado-Rives J, et al. OPLS all-atom force field for carbohydrates[J]. Journal of Computational Chemistry, 1997, 18(16): 1955-1970
[11]Kony D, Damm W, Stoll S, et al. An improved OPLS-AA force field for carbohydrates[J]. Journal of Computational Chemistry, 2002, 23(15): 1416-1429
[12]Daura X, Gademann K, Jaun B, et al. Peptide folding: when simulation meets experiment[J]. Angewandte Chemie International Edition, 1999, 38(1-2): 236-240
[13]Wang X Y, Zhang L, Wei X H, et al. Molecular dynamics of paclitaxel encapsulated by salicylic acid-grafted chitosan oligosaccharide aggregates[J]. Biomaterials, 2013, 34(7): 1843-1851
[14]Fedorov M V, Goodman J M, Kolombet V V, et al. Conformational changes of trialanine in sodium halide solutions: An in silico study[J]. Journal of Molecular Liquids, 2009, 147(1): 117-123.
The Complexation Behavior of Amylose and α-Linoleic Acid by Molecular Dynamics Simulations
Feng Tao1Wang Xu1Zhuang Haining2Feng Chen3Bruce R Hamaker3Osvaldo Campanella3Wang Kai4
(School of Perfume and Aroma Technology, Shanghai Institute of Technology1, Shanghai 201418)(Institute of Edible Fungi, Shanghai Academy of Agricultural Sciences,National Engineering Research Center of Edible Fungi2, Shanghai 201403)(Whistler Center for Carbohydrate Research, Department of Food Science, Purdue University3, IN 47906-2009, USA)(R&D Center, China Tobacco Yunnan Industrial Co., Ltd4, Kunming 650231)
The amylose could form a thermodynamically stable complex with α-linoleic acid. In order to study the mechanism that α-linoleic acid affecting the configuration of amylose; the favorable configuration of amylase and α-linoleic acid complex; and the changes of hydrogen bonds during the complexation process, a long time (500 ns) all-atom molecular dynamics (MD) simulation has been performed at the temperature of 373 K. In both the presence and absence of α-linoleic acid, a large-scale order-to-disorder conformational transition happened within 500 ns was observed, suggesting the weak coupling between amylose and α-linoleic acid. The results of the atom root mean square deviation (RMSD), the axial distance between α-linoleic acid and amylose, and the increment of hydrogen bonds, all supported a common conclusion that amylose-α-linoleic acid complex was formed by thermodynamically spontaneous behavior.
molecular dynamics simulation, amylose, α-linoleic acid
TS231
A
1003-0174(2016)04-0035-06
国家自然科学基金 (31000794)
2014-08-11
冯涛,男,1978年出生,教授,食品风味化学
猜你喜欢
同位素(2022年6期)2022-12-30
物理学报(2022年10期)2022-06-04
中国农业科学(2021年7期)2021-04-21
唐山师范学院学报(2020年6期)2020-04-16
中国粮油学报(2019年4期)2019-07-12
唐山师范学院学报(2019年3期)2019-06-18
中国洗涤用品工业(2019年4期)2019-05-11
中国粮油学报(2016年10期)2016-12-26
中国粮油学报(2015年5期)2015-02-06
中国药业(2014年12期)2014-06-06
