
铜表面硅烷裂解的第一性原理研究
2017-02-01 01:15王铎李佳
当代化工 2017年12期
王铎,李佳
铜表面硅烷裂解的第一性原理研究
王铎,李佳*
(广东省热管理工程与材料重点实验室, 清华大学深圳研究生院,广东 深圳 518055)
通过基于密度泛函理论的第一性原理计算,研究硅烷(SiH4)在Cu(111),Cu(100),Cu(110)表面的裂解过程,确定了4个反应步骤的所有反应物、生成物以及中间产物的稳定吸附位置。并且从热力学角度分析每一步反应的驱动力,得出硅烷裂解为放热反应以及硅烷的第一步裂解(SiH4→SiH3+H)最容易发生。因此我们推测在化学气相沉积过程中,在铜表面很难观测到硅烷分子的存在。
硅烷;铜表面;裂解;第一性原理
石墨烯具有极其优异的性质,比如极高的力学强度和热导率[1],极好的导电性[2],以及室温量子霍尔效应[3]等。但是,本征石墨烯的导带和价带在布里渊区Gamma点呈Dirac锥形接触,使石墨烯成为一种具有零带隙的半导体。即使是稳定的零带隙材料,由于无法实现逻辑电路的开关,也对石墨烯的应用造成了极大的限制。因此,对石墨烯带隙的调控也越来越受到人们的重视。其中一个比较常见的方法就是异质原子的掺杂[4-9]。
化学气相沉积法(CVD)是一种有望大规模、高质量制备石墨烯的一种实验方法。可以控制生产过程中异质原子的掺入来形成有效地替位掺杂[10],从而对石墨烯的性能进行调控。目前已经有研究着眼于模拟化学气相沉积法(CVD)制备石墨烯这一化学过程[11]。
在实验和理论计算领域,已经有学者对石墨烯的正常生长过程进行了深入的探讨[11-16],但是对于异质原子的掺杂过程,尤其是CVD方法下生长替位掺杂石墨烯的过程还没有被深入研究。本文就是我们以第一性原理的方法对硅烷裂解过程进行模拟,目的是与甲烷裂解相比较,以了解硅原子在CVD制备石墨烯的常用基底上的行为,为之后我们模拟CVD法制备硅掺杂石墨烯的相关研究作准备。
1 计算方法
我们的所有计算都在VASP软件包[17,18]上进行,采用基于密度泛函理论(DFT)的投影缀加波赝势,广义梯度近似(GGA)的PBE交换关联泛函,计算过程中考虑范德华力(DFT-D2)。我们建立了4层4×4的Cu(111),Cu(100),Cu(110)衬底模型,真空层15 Å。固定底部两层以保证这两层原子与铜的体相结构一致,优化其它所有原子的位置。截断能为300 eV,力收敛精度为0.01 eV/Å,选取5×5×1的K点网格。
2 结果与讨论
2.1 建立模型
我们首先优化了面心立方Cu单胞的晶格常数。采用扫点法——取不同晶格常数的原胞,做静态计算以优化其能量。最终比较所有不同晶格常数下单胞的能量,得到的能量最低结构所对应的数值即为我们计算的该体系下面心立方金属Cu的晶格常数,如图1所示。得到Cu晶格常数为3.57 Å,这与实验上得到的数值3.61 Å相差0.04 Å,约1.13%,所以可以认为我们的计算合理、准确(图1)。

图1 面心立方铜原胞的晶格常数
继续对晶格常数为3.57 Å的Cu单胞建立超胞模型。面心立方Cu金属的三种金属表面Cu(111),Cu(100),Cu(110)的模型如图2所示。
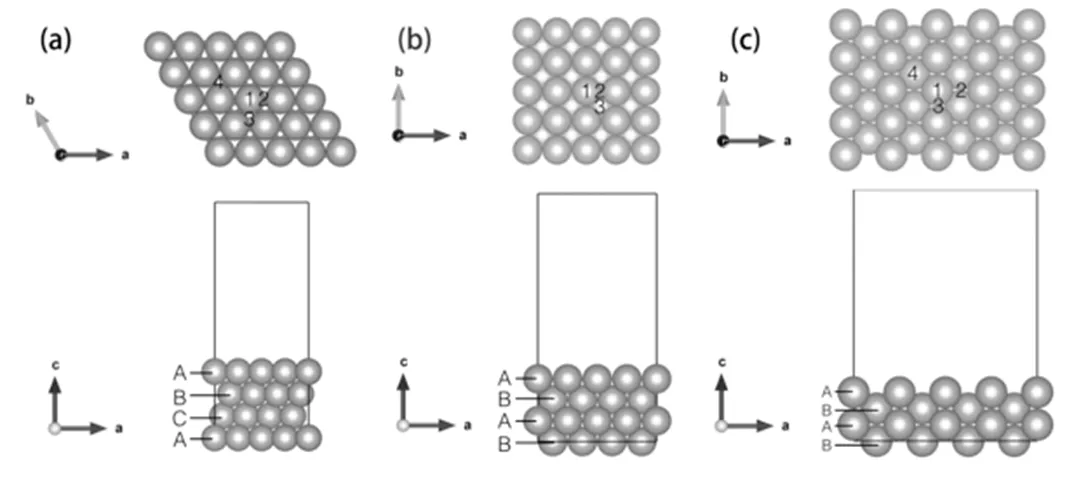
图2 金属衬底模型
(a)Cu(111)表面模型;(b)Cu(100)表面模型;(c)Cu(110)表面模型
三种不同指数的衬底具有各自不同的特征,如图2(a)所示,Cu(111)为面心立方金属Cu的密排面,由顶视图可以看出该衬底为六方结构,a、b轴之间的夹角为120。,基矢=,该表面的密排程度最高,表面最近邻原子之间距离为2.56 Å,面上的每个铜原子与6个原子相连。从侧视图可以看到Cu(111)表面的堆垛为ABCABC……的周期性结构。图2(a)顶视图中,不同的数字标注表示该表面的不同的高对称位置:1为顶位,2为桥位,3、4为空隙位,其中3为密排六方空隙,4为面心立方空隙。根据这两种空隙的命名方式我们很好理解,3位置正对着衬底的第二层原子,而4位置正对第三层原子。在研究表面吸附问题时。Cu(100)表面为ABABAB……堆垛,四方结构,表面上共有三个不同的高对称位置:1顶位,2桥位,3空隙位,如图2(b)所示。在本论文所研究的三个表面中,Cu(100)表面密排程度略低于Cu(111)表面,最近邻原子间距为2.56 Å,但与Cu(111)不同的是,表面上每个铜原子只有4个原子与之相连。图2(c)为Cu(110)表面,ABABAB……堆垛,三个表面中,Cu(110)的密排程度最低,这就导致表面结构相对于另外两个表面有不同的特点。我们在图中标出了表面上几个不同的高对称点:1顶位,2、3桥位,造成这两个桥位不同的原因在于表面相对稀疏的原子排布和层间ABAB的堆垛方式,位置2是位于相距比较远的两原子之间的桥,3是近邻原子间的桥位,Cu(110)表面最近邻两原子间距为同样是2.56 Å,但此时仅有两个原子,在平面上沿图中b方向相连。
对这三种表面进行结构优化后,我们分别计算了三种表面的表面能:

式中:E—计算得到的表面结构的能量,eV;
N—总原子数;
—表面面积,eV/nm2。
据此,我们将计算得到三个表面的表面能数值列于表1。

表1 金属铜三个不同指数表面的表面能
表面能与其密排程度直接相关,密排程度越高,表面原子的配位数越高,未饱和电子数越少,结构越稳定,从而表面能越低。
2.2 硅烷及其反应中间产物在金属表面的吸附
硅烷的裂解包括4个反应过程:
SiH4→SiH3+H (2)
SiH3→SiH2+H (3)
SiH2→SiH+H (4)
SiH→Si+H (5)
在反应的过程中共有SiH4、SiH3、SiH2、SiH、Si、H这6种反应物或中间产物。我们首先计算了这6种分子在金属衬底表面吸附的最稳定结构,并对每个结果进行具体分析。
2.2.1 SiH4在Cu(111),Cu(100)和Cu(110)表面上的吸附
如图3所示,SiH4结构吸附于三种铜表面上的结构是比较相似的,两个H原子位于表面铜原子的正上方,Si与靠近金属表面的两个H原子形成的键角为150。,与远离表面的两个氢原子形成的键角为109。(这个角度与硅烷在真空中的键角一致),Cu-H键长约等于1.62 Å。这三个结构中唯一的差别在于Si原子与靠近金属表面的H原子的键长,对应Cu(111),Cu(100),Cu(110)三种表面分别为1.67,1.69,以及1.70 Å,相对于硅烷在真空中的Si-H键长(1.50Å)有显著的增大。可见SiH4与衬底有明显的相互作用。计算得到三种情况的吸附能分别为:0.63,0.79以及0.89 eV。相对于CH4吸附于金属表面的情形[11],SiH4分子与金属表面明显有较强的相互作用。

图3 SiH4在三种铜基底上的吸附
(a)Cu(111)表面; (b)Cu(100)表面; (c)Cu(110)表面
2.2.2 SiH3在Cu(111),Cu(100)和Cu(110)表面上的吸附
图4 是SiH3在三种铜衬底表面的吸附。
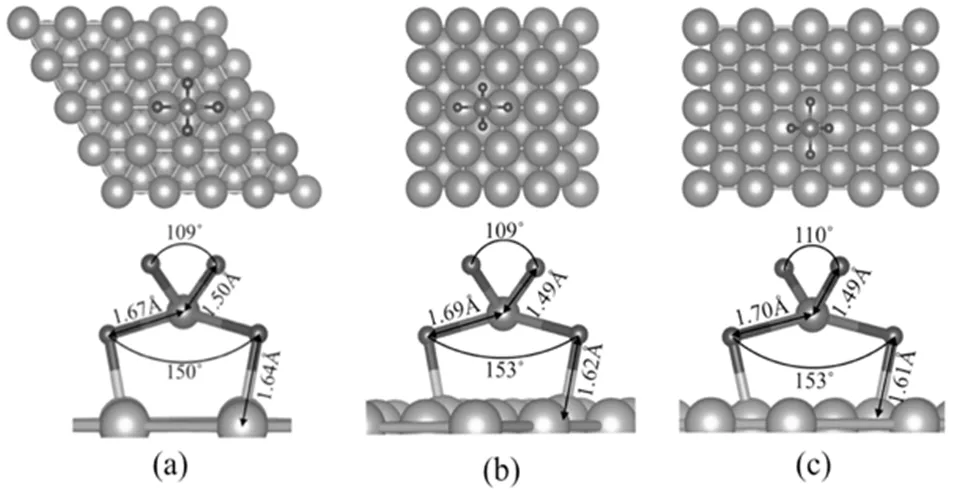
图4 SiH3在三种铜基底上的吸附
(a)Cu(111)表面; (b)Cu(100)表面; (c)Cu(110)表面
可以看到吸附于Cu(111)表面的结构与其它两个结构区别较大:两个氢原子远离表面,另一氢原子在表面一铜原子的正上方,Cu-H键长为1.75 Å。在Cu(100)和Cu(110)表面上,SiH3的三个氢原子中,两个氢原子处于表面铜原子的正上方,Cu-H键长分别为1.67,1.68 Å,另一个氢原子的Si-H键长与真空中SiH3分子的一致(1.5 Å)。同时Si-Cu键长约为2.45 Å。这三个结构的一个共同点是Si原子的位置,都在空隙位。SiH3分子在三种表面的吸附能分别为2.91,3.25,3.30 eV。同样与表面的密排程度呈负相关,即表面密排程度越大,吸附能越低。归根到底,吸附能与衬底及分子的结构密切相关。
2.2.3 SiH2在Cu(111),Cu(100)和Cu(110)表面上的吸附
如图5所示,SiH2分子在三种铜表面的吸附状态,三种稳定吸附结构是一致的,Si原子处于空隙位(Cu(111)表面上为面心立方空隙)。虽然靠近表面的H原子状态看起来略有差异——与Cu(111)、Cu(100)表面上H原子位于Cu原子的顶位不同,Cu(110)上H原子略微偏离Cu顶——但进一步分析我们发现,所有Cu-H键长是一致的,都在1.70Å左右。另一方面, Cu-Si键长略有区别,对Cu(111),Cu(100),Cu(110)表面分别为2.28,2.37,2.44 Å,与SiH3结构相比,Cu(111)及Cu(100)表面上的Cu-Si键长略有减小。也可以从另一个角度来看,随着表面密排程度增大,Cu-Si键长有一定程度的减小。我们分析原因在于额外H离子的脱去,导致Si原子在密排面倾向于降低位置,以增加分子-金属表面的相互作用。SiH2分子在这三种表面的吸附能分别为3.03,3.55,3.66 eV。总体仍然符合密排程度越低,表面越活跃,分子-表面相互作用更强,从而吸附能更大这一规律。

图5 SiH2在三种铜基底上的吸附
(a)Cu(111)表面; (b)Cu(100)表面; (c)Cu(110)表面
2.2.4 SiH在Cu(111),Cu(100)和Cu(110)表面上的吸附
如图6所示,SiH分子吸附于三个表面上时,Si原子处于空隙位(对于Cu(111)表面为面心立方空隙),而H原子的位置略微有些差别,Cu(111)、Cu(100)表面上时,H原子位于表面金属原子的顶位,Cu(110)面时H只与Si原子相连,远离金属。这样就造成Si-H键长的不同,前两者为1.62 Å,Cu(110)仍保持气态SiH的结构,键长为1.51 Å,我们分析造成这一结构区别的原因在于,Cu(110)表面密排程度低,当氢原子同样处于衬底金属原子顶位时,Si、H原子因为距离过长无法成键。SiH分子在这三种不同表面的吸附能分别为3.89,4.83,以及4.90 eV。

图6 SiH在三种铜基底上的吸附
(a)Cu(111)表面; (b)Cu(100)表面; (c)Cu(110)表面
2.2.5 Si、H原子分别在Cu(111),Cu(100)和Cu(110)表面上的吸附
Si、H原子的吸附结果比较一致,全部位于表面的空隙位置,这样可以最大化被吸附原子与表面原子的配位数。Si原子的吸附能分别为5.01,5.80,6.00 eV;H原子吸附能分别为3.64,3.62,3.57 eV。这里Cu(111)的两个不同空隙位对能量影响较小,比如Si吸附于这两种空隙位的能量差约为44 meV,所以两种空隙位的影响可以忽略不计。
2.2.6 SiH_(=1,2,3,4)和H在三种铜表面的吸附总结
综上,我们将硅烷裂解过程中所有反应物、生成物以及中间产物的吸附情况总结于表2。

表2 硅烷裂解的反应物、中间产物以及生成物在铜三个表面上的吸附能
整体来看,SiH4分子的吸附能最低。并根据硅烷裂解的反应公式可以得到,每一步裂解之后的吸附能明显高于反应前,举例来说,Cu(111)表面的第一步反应为:
SiH4→SiH3+H (6)
反应前的吸附能时0.63 eV,反应后SiH3和H的吸附能之和为6.55 eV,整体为放热反应,我们将生成物的总吸附能与反应物的吸附能相减,把这一数值定义为反应的驱动力,以第一步反应为例:
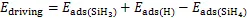
式中:driving—驱动力,eV;
ads—吸附能,eV。
据此,我们将每个反应在三个表面上的驱动力总结于表3。

表3 铜三种衬底上每一步裂解反应的驱动力
正数代表发生放热反应,数值的大小代表反应驱动力的强弱。由表中数据可以直观地看出硅烷裂解的所有过程均为放热反应。纵向来看,对于所有表面上的硅烷裂解,我们可以得到结论:第一步反应的驱动力最大,第二步驱动力最小。
3 总结
本文运用基于密度泛函理论的第一性原理方法,系统地研究了硅烷在铜的三种不同指数表面(Cu(111)、Cu(100)、Cu(110))上的裂解。首先计算了SiH4及其反应中间产物在表面上的稳定吸附位点,结合Si-H、Cu-H以及Cu-Si键长分析了每种吸附结构。并且从热力学角度计算了发生反应的驱动力。我们将得到的结果与甲烷在铜表面的裂解12相比较,得到以下的三个结论:首先,所有反应中间产物及生成物都倾向于吸附在表面的空隙位;第二,硅烷裂解的每一步反应都是放热反应;第三,硅烷在每个铜表面裂解的驱动力第一步最大,第二步最小,据此我们推测在CVD实验中很难在铜表面上观察到SiH4结构,而有较大数目SiH3分子在铜表面。
[1]Lin Y, Guo X, Chemical modification of graphene and its applications[J]. Huaxue Xuebao, 2014, 72: 277-288.
[2]Zomer P J, Dash S P, Tombros N, et al. A transfer technique for high mobility graphene devices on commercially available hexagonal boron nitride[J]. Applied Physics Letters, 2011, 99(23): 232104.
[3]Novoselov K S, Fal V I, Colombo L, et al. A roadmap for graphene[J]. Nature, 2012, 490(7419): 192-200.
[4]Lv R, Li Q, Botello-Méndez A R, et al. Nitrogen-doped graphene: beyond single substitution and enhanced molecular sensing[J]. Scientific Reports, 2012, 2: 586.
[5]Han J, Zhang L L, Lee S, et al. Generation of B-doped graphene nanoplatelets using a solution process and their supercapacitor applications[J]. ACS Nano, 2012, 7(1): 19-26.
[6]Zhao L, Levendorf M, Goncher S, et al. Local atomic and electronic structure of boron chemical doping in monolayer graphene[J]. Nano Letters, 2013, 13(10): 4659-4665.
[7]Denis P A. Band gap opening of monolayer and bilayer graphene doped with aluminium, silicon, phosphorus, and sulfur[J]. Chemical Physics Letters, 2010, 492(4): 251-257.
[8]Gao H, Liu Z, Song L, et al. Synthesis of S-doped graphene by liquid precursor[J]. Nanotechnology, 2012, 23(27): 275605.
[9]Some S, Kim J, Lee K, et al. Highly air‐stable phosphorus‐doped n‐type graphene field‐effect transistors[J]. Advanced Materials, 2012, 24(40): 5481-5486.
[10]Lv R, dos Santos M C, Antonelli C, et al. Large‐Area Si‐Doped Graphene: Controllable Synthesis and Enhanced Molecular Sensing[J]. Advanced Materials, 2014, 26(45): 7593-7599.
[11]Zhang W, Wu P, Li Z, et al. First-principles thermodynamics of graphene growth on Cu surfaces[J]. The Journal of Physical Chemistry C, 2011, 115(36): 17782-17787.
[12]Kidambi P R, Bayer B C, Blume R, et al. Observing graphene grow: catalyst–graphene interactions during scalable graphene growth on polycrystalline copper[J]. Nano Letters, 2013, 13(10): 4769-4778.
[13]Li X, Magnuson C W, Venugopal A, et al. Graphene films with large domain size by a two-step chemical vapor deposition process[J]. Nano Letters, 2010, 10(11): 4328-4334.
[14]Sutter P W, Flege J I, Sutter E A. Epitaxial graphene on ruthenium[J]. Nature Materials, 2008, 7(5): 406-411.
[15]Tao Y, Xue Q, Liu Z, et al. Tunable hydrogen separation in porous graphene membrane: first-principle and molecular dynamic simulation[J]. ACS Applied Materials & Interfaces, 2014, 6(11): 8048-8058.
[16]Aktürk O Ü, Tomak M. Adsorption of RuSex (= 1–5) cluster on Se-doped graphene: First principle calculations[J]. Applied Surface Science, 2015, 347: 808-815.
[17]Kresse G, Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set[J]. Computational Materials Science, 1996, 6(1): 15-50.
[18]Kresse G, Hafner J. Ab initio molecular dynamics for liquid metals[J]. Physical Review B, 1993, 47(1): 558.
First Principles Study of Silicane Decomposition on Copper Surface
WANG Duo, LI Jia
(Guangdong Provincial Key Laboratory of Thermal Management Engineering and Materials, Graduate School at Shenzhen, Tsinghua University, Guangdong Shenzhen 518055, China)
First principles calculations using density functional theory were performed on the decomposition process of silicane on three different copper surfaces: Cu(111), Cu(100) and Cu(110). The stable adsorption configurations of reactants, products and intermediates on copper surface in four reaction processes were determined. In consideration of thermodynamics, we found that all reactions are exothermic and the first reaction step (SiH4→SiH3+H) is the easiest reaction. So it’s predicted that, in the process of chemical vapor deposition, SiH4is difficult to exist on the copper surface.
Silicane;Copper surface;Decomposition;First principles
TB303
A
1671-0460(2017)12-2403-04
深圳市孔雀计划创新创业基金,项目号:KQCX20140521161756227。
2017-05-19
王铎(1990-),男,内蒙古乌海市人,清华大学在读硕士生,研究方向:第一性原理计算。E-mail:wangd14@mails.tsinghua.edu.cn。
李佳(1980-),男,副教授,博士,研究方向:计算凝聚态物理和材料设计研究。E-mail:li.jia@sz.tsinghua.edu.cn。
猜你喜欢
中国交通信息化(2021年1期)2021-06-11
陶瓷学报(2020年5期)2020-11-09
纺织科学与工程学报(2020年1期)2020-06-12
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中国科技信息(2016年6期)2016-08-31
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16
中学化学(2015年8期)2015-12-29
中国科技信息(2015年24期)2015-11-07
中国科技信息(2015年23期)2015-11-07
上海塑料(2015年3期)2015-02-28
