
脊髓性肌萎缩伴呼吸窘迫1型不伴呼吸衰竭1例报告
2017-04-26 02:50孙龙妹
临床儿科杂志 2017年3期
郭 莉 孙龙妹 刘 芳
中国人民解放军白求恩国际和平医院新生儿科(河北石家庄 050082)
脊髓性肌萎缩伴呼吸窘迫1型不伴呼吸衰竭1例报告
郭 莉 孙龙妹 刘 芳
中国人民解放军白求恩国际和平医院新生儿科(河北石家庄 050082)
目的探讨脊髓性肌萎缩伴呼吸窘迫1型(SMARD1)的诊断和鉴别诊断。方法回顾分析1例确诊为SMARD1女性患儿的临床资料、基因检测结果及随访,并复习相关文献。结果患儿因羊水过少剖宫产,生后吃奶、反应欠佳转入新生儿科,诊断“新生儿脓毒症、感染性休克、弥散性血管内凝血、非典型化脓性脑膜炎”,经治疗1个月后出院;生后2个月出现踝关节挛缩,肝功能异常、心肌受损;6个月时发现肌张力明显减低、运动发育落后;8个月时行SMA相关基因检测结果阴性;9个月时行周围神经病panel基因检测发现IGHMBP2基因存在2个杂合突变,即exon8 c.1061-2A>G和exon12 c.1708C>T,分别来自父亲和母亲,其中位点exon12 c.1708C>T已有文献报道与疾病相关,另1个为剪切突变,结合临床确诊为SMARD1。患儿现2岁,反复合并呼吸道感染,但尚未出现呼吸窘迫或呼吸衰竭。结论SMARD1的临床表型复杂多样,该例患儿为经基因诊断病例。
脊髓性肌萎缩伴呼吸窘迫1 型; 呼吸衰竭; 膈肌麻痹
脊髓性肌萎缩伴呼吸窘迫1型(spinal muscular atrophy with respiratory distress type 1,SMARDl)是一种极为罕见的常染色体隐性遗传病。由于常染色体11 q 13上的免疫球蛋白μ结合蛋白2(immunoglobulinμ-binding protein 2,IGHMBP2)基因发生突变[1],导致脊髓前角α-运动神经元和脊髓背侧神经根细胞发生不可逆的退行性变性[2],最为突出的特征是婴儿期在毫无预兆下突发不可逆性膈肌麻痹导致呼吸窘迫、呼吸衰竭,危及生命[3]。但少数患儿不以膈肌麻痹或呼吸功能障碍为首发症状,国外已相关病例报道[4-6],而国内尚未见报道。现对我院确诊为SMARD1不伴呼吸衰竭患儿的病史进行回顾性分析,并结合相关文献复习,探讨疾病的临床表现、诊断、鉴别诊断和治疗进展,借此期望能引起临床儿科医师对该病的认识,提高对遗传性疾病的重视。
1 临床资料
患儿,女,G3P3,胎龄39+6周,因羊水过少行剖宫产娩出,出生体质量2 200 g,头围29 cm,身长49 cm;Apgar评分不详,否认出生时窒息、抢救史,生后一般情况好。父母体键,非近亲婚配;哥哥现11岁,体键。母亲第2胎为女孩,4个月时突发呼吸困难,经抢救无效死亡;此次孕7个月时产检提示胎儿宫内生长发育迟缓。
患儿生后第2天,因吃奶欠佳1天转入新生儿科。当时体温37.7℃,刺激反应欠佳,哭声弱;全身皮肤干燥,躯干及四肢皮肤发花,四肢末梢凉。血常规白细胞(WBC)22.27×109/L,血小板(Plt)30×109/L;血涂片杆状核/中性粒细胞 22%;天冬氨酸氨基转移酶88 U/L,肌酸激酶396 U/L,肌酸酶同工酶67 U/L,乳酸脱氢酶798 U/L;纤维蛋白原0.53 g/L,D-二聚体1.62 mg/L;脑脊液常规及生化结果无异常;优生四项(TORCH)阴性;血培养无细菌及真菌生长;血、尿遗传代谢性疾病筛查阴性。诊断为“新生儿脓毒症(临床型)、感染性休克、弥散性血管内凝血(早期)、非典型化脓性脑膜炎”,给予补充凝血因子,能量合剂营养支持,超小剂量低分子肝素钠皮下注射抗凝,联合抗感染治疗,逐步恢复正常。患儿住院期间吃奶欠佳,吸吮弱,体质量、头围增长缓慢,血氨、乳酸正常。头颅CT示脑沟脑裂显示欠佳、双侧侧脑室较小。予胞磷胆碱钠、神经节苷脂钠营养脑细胞,吃奶情况逐渐好转,经综合治疗1个月病情平稳出院。
生后2个月,患儿足背屈角增大,再次就诊。当时体格检查:营养中等,精神反应好,眼神可追物,有试抬头动作,足背屈角90°,有阻力,踏步反射可引出,足尖着地,双足交叉,双侧髋关节外展稍差,下肢肌张力稍高,上肢肌张力正常;心肺无异常;腹软,肝肋下2.5 cm,质软,边锐,脾肋下1 cm,肠鸣音正常。实验室检查:谷氨酸氨基转移酶113 U/L,天冬氨酸氨基转移酶142 U/L,肌酸激酶306 U/L,肌酸激酶同工酶102 U/L。心电图无异常。头颅磁共振成像(MRI)未见明显异常。给予保肝、营养心肌治疗,1周后建议前往专业机构行康复训练。生后6个月,因四肢肌力、肌张力减低,运动发育落后4个月就诊于白求恩国际和平医院小儿康复科门诊。婴儿神经系统测试国际量表(Infant Neurological International Battery,INFANIB)评估示66分。足背屈角90°(偏大),站立位不能承重,坐位身体前倾明显,俯卧位不能抬头,拉起头后仰、肩滞后,主动抓握准确性差,能逗笑,偶能笑出声,会吃手,双手能合于中线,运动落后于同龄儿。当时体格检查示体温36.4℃,脉搏100次/min,呼吸34次/min,身长63.5 cm,体质量6.2 kg(均低于第3百分位数);神清,营养欠佳,认知、运动发育均落后于正常同龄儿;全身皮肤黏膜无皮疹、色素沉着,无咖啡牛奶斑,全身浅表淋巴结未扪及,易出汗,掌心为著;双侧眼睑无下垂,眼球运动无受限,双侧瞳孔等大等圆,对光反射灵敏;伸舌居中,无舌尖震颤;两侧鼻唇沟等深;颈软,无抵抗,颈肌无力,抬头困难;胸廓对称无畸形,双肺呼吸音清;心律齐,未闻及杂音;腹部平软,肝脾肋下未触及;脊柱无畸形,双侧踝关节挛缩,呈伸直状态,双足不能背屈或跖屈,关节无红肿、触痛或叩击痛;四肢肌力、肌张力明显减低,肌肉未见萎缩性改变;膝、跟腱反射均消失,踝阵挛阴性,双侧巴氏征、克氏征、布氏征均阴性。实验室检查示丙氨酸氨基转移酶87.4 U/L,门冬氨酸氨基转移酶142.7 U/L,磷酸肌酸激酶381 U/L,磷酸肌酸激酶同工酶117 U/L,乳酸脱氢酶716 U/L;游离三碘甲状腺原氨酸(FT3)7.48 pmol/L,三碘甲状腺原氨酸4.13 pmol/L。外周淋巴细胞染色体分析示46,XX。超声心动图示心内结构未见明显异常。颅脑MRI示双侧额部轻度外部性脑积水,右侧乳突可疑炎性改变。胸片示两肺炎症性改变(图1)。

图1 患儿胸片
患儿8个月时,经医院医学伦理委员会批准,父母知情同意,抽取患儿和其父母的外周静脉血各2 mL,行基因检测。脊髓性肌萎缩(spinal muscular atrophy,SMA)相关基因筛查结果阴性。周围神经病panel基因测序示患儿IGHMBP2基因有2个杂合突变,即exon8 c.1061-2A>G和exon12 c.1708C>T,分别来自父亲和母亲,其中位点exon12 c.1708C>T已有文献报道[7],为SMA伴呼吸窘迫1型的致病突变位点,另一个位点为剪切突变,提示患者为SMARD1(图2)。入院后给予葡醛内酯钠、肝泰乐、维生素C保肝治疗,小儿肢体康复训练、感觉统合训练、水疗等促通运动发育,住院期间合并肺炎,给予雾化、吸痰、抗感染治疗;行肌电图检查示神经源性损伤。经半年的综合康复训练,运动发育情况未见明显好转,家长要求出院行家庭康复训练。
患儿现2岁,长期卧床,营养状况差,智力、运动发育情况明显落后,只会说“爸爸、妈妈”等简单词汇,与人交流差,不会抬头、翻身、坐、爬等,四肢肌力、肌张力降低,双上肢肌力近端4级,远端3级,双下肢肌力近端3级,远端2级,肌肉可见明显萎缩,膝关节及肘关节无明显变形,脊柱无侧弯。患儿于生长过程中因反复合并呼吸道感染住院治疗,但一直未发生呼吸窘迫和呼吸衰竭。
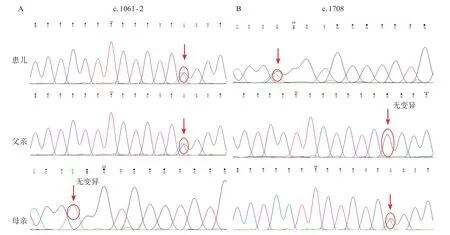
图2 患儿及其父、母IGHMBP2 基因分析结果
2 讨论
SMARD 1为常染色体11 q 13上的IGHMBP2基因突变所致的一种极为罕见的隐性遗传性疾病,又名远端型脊肌萎缩症1型(distal spinal muscular atrophy 1,DSMA1)或远端遗传性运动神经病6型(distal hereditary motor neuropathies type 6,dHMN6)。SMARD 1确切的患病率尚不清楚,因为约1%的SMA伴膈肌麻痹患者被诊断为早发型SMA[8]。目前,SMARD1的详细发病机制仍不清楚,但IGHMBP2基因突变致脊髓前角α运动神经元变性是疾病的基础。SMARD1的主要临床特点为婴儿期(常为出生6周~6个月)无明显诱因出现逐渐进展的对称性肢体远端肌无力(尤其是下肢)和不可逆性膈肌麻痹导致呼吸衰竭,需机械通气维持呼吸。一般预后较差,大部分患儿于10岁内死亡[9],少数在人工辅助呼吸下存活至20岁以上[10,11]。
在解旋酶超家族1(SF1)成员中,IGHMBP2是一种广泛表达的ATP酶/解旋酶,含有15个外显子,编码993个氨基酸, 包含解旋酶结构域、单链核酸结合区域以及1个锌指基序,参与DNA解旋或RNA合成,在细胞核中参与调控转录及tRNA的加工[12,13]。SMARD1的基因突变以单一碱基的置换、插入、缺失等点突变为主,以复合杂合突变和纯合突变为主,至今已有60余例SMARD1被报道[3],发现了140多个IGHMBP2基因突变位点。除IGHMBP2基因的第4外显子,其余外显子均有突变致病报道。本例患儿基因测序中发现IGHMBP2基因存在exon8 c.1061-2A>G和exon12 c.1708C>T的杂合突变,其中exon12 c.1708C>T位点已有文献报道[7]。
多数SMARD1患儿出生时四肢肌力、肌张力基本正常,于生后6周~6个月内出现对称性远端肌无力、肌萎缩,以下肢为著,逐渐向肢体近端及躯干蔓延,伴突发性膈肌麻痹致呼吸窘迫,需气管插管、机械通气辅助呼吸维持生命。多数于生后2个月左右出现踝关节挛缩,随着病情进展,逐渐出现膝关节、肘关节、胸廓及脊柱的变形,上、下肢指节可见特征性脂肪垫[14],腱反射减弱或消失。其中最为突出的特征性表现是在毫无预兆下突发膈肌麻痹致呼吸窘迫危及生命,胸部X线下可见特征性膈肌膨出,可为单侧或双侧,多见于右侧,可能与肝脏对麻痹的膈肌有一定向上的压力有关。SMARD1主要累及周围运动神经和呼吸系统,此外还可以累及中枢神经、感觉神经、自主神经以及其他脏器的功能异常。累及中枢神经系统,表现为精神发育迟缓、癫痫样发作、面部表情肌受累和舌肌震颤等;累及感觉神经表现为痛觉下降等;自主神经功能紊乱表现为体温、血压不稳、心率失常、易出汗、神经源性膀胱等。虽然目前尚无SMARD1患儿心肌受损的相关报道,但无义介导的mRNA降解(NMD)小鼠模型中可观察到心脏受损表现,如心率失常、扩张型心肌病等,所以是否合并心肌受损需进一步研究。孕期可出现宫内发育异常等非特异性表现,如胎动减少、羊水过少、早产、小于胎龄儿等。
本例患儿孕7个月时产检提示宫内生长发育迟缓,出生时因羊水过少剖宫产,为足月小样儿。生后第2天诊断新生儿脓毒症(临床型)、感染性休克、弥散性血管内凝血(早期)、非典型化脓性脑膜炎等;生后2个月时出现踝关节挛缩、足尖着地;生后6个月时肌张力明显减低、运动发育明显落后,腱反射消失,肌电图提示神经源性损伤,怀疑脊髓性肌萎缩,行SMA相关基因检测结果为阴性。但患儿症状仍不除外周围神经性疾病,扩大基因筛查范围发现IGHMBP2基因存在2个杂合突变,进一步验证患儿父母的IGHMBP2基因,明确父母为该位点的杂合突变携带者,结合临床,最终确诊为SMARD1。患儿现2岁,多次因呼吸道感染住院治疗,多次行胸部X线平片均未发现膈肌抬高,至今未出现呼吸窘迫、呼吸衰竭。
SMARD1患儿临床表型存在较大的差异性,即使同胞兄弟姐妹存在相同的突变位点临床表现也不完全相同[15]。本例患儿与已故同胞姐姐携带相同突变位点,姐姐于生后6个月时突发呼吸困难,经抢救无效死亡,本例患儿2岁仍未出现呼吸功能不全症状。Hamilton等[10]报道1例21岁女性基因确诊为SMARD1,合并稳定型肌无力,正常的认知能力,能适应社会集体化生活,仅夜间需要机械通气维持呼吸。Xinghua等[16]报道1例患儿4个月时出现四肢远端肌无力、肌萎缩,但直到生后54个月仍未出现呼吸功能不全症状。近期研究表明,残留的IGHMBP2蛋白水平与疾病的严重程度有关。即残留的IGHMBP2蛋白量越多,患儿的发病时间越迟,保留的运动功能越多[17]。
SMARD1目前尚无统一的诊断标准,血清心肌酶谱、肌电图、肌肉活检和IGHMBP2基因检测均有助于诊断和鉴别诊断。临床上出现呼吸衰竭合并以下症状或体征时高度怀疑SMARD1[18]:①呼吸衰竭在出生后6周~6个月出现;②膈肌麻痹;③远端性肌无力;④宫内生长受限;此时需行特异性IGHMBP2基因检测明确诊断。然而在分析基因检测结果时,应注意基因突变的多态性、突变类型及基因突变对编码蛋白功能的影响,基因检测结果必须结合临床表现。
因SMARD1临床表型的多样性,极易发生误诊或漏诊,需与以下常见遗传性疾病相鉴别。①脊髓性肌萎缩1型(SMA1):常染色体5q13上的运动神经存活基因1(SMN1)突变致隐性遗传病,生后6个月内起病,表现为对称性四肢近端肌无力,疾病后期可出现呼吸衰竭,基因分析可明确诊断[19]。②先天性肌强直性肌营养不良1型:常染色体19q13.32上编码强直性肌营养不良蛋白激酶(DMPK)基因突变致遗传性疾病,孕期表现为胎动减少,出生时即表现为松软儿,吸吮、吞咽无力,常伴足、关节畸形,早期肋间肌变薄,膈肌受累,出现呼吸困难。因患儿1岁内多无肌强直表现,肌电图无强直电位,容易误诊,检测DMPK基因3’非编码区内的核苷酸(CTG)重复次数大于50个以上可以诊断[20]。
目前SMARD1尚无有效的治疗方法,主要以保持呼吸道通畅、机械通气辅助呼吸灯对症治疗延长患儿的生存期,改善生活质量,但干细胞移植和基因疗法被看作是潜在有效的治疗方案[21]。IGHMBP2基因检测是目前诊断SMARD1的唯一可靠方法,可通过产前诊断评估生育患儿的遗传风险。
[1] Grohmann K, Schuelke M, Diers A, et al. Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1 [J]. Nat Genet, 2001, 29(1):75-77.
[2] San Millan B, Fernandez JM, Navarro C, et al. Spinal muscular atrophy with respiratory distress type 1 (SMARD1). Report of a Spanish case with extended clinic pathological follow-up [J]. Clin Neuropathol, 2016, 35(2):58-65.
[3] Porro F, Rinchetti P, Magri F, et al. The wide spectrum of clinical phenotypes of spinal muscular atrophy with respiratory distress type 1: a systematic review [J]. J Neurol Sci, 2014, 346(1-2): 35-42.
[4] Stalpers XL, Verrips A, Poll-The BT, et al. Clinical andmutational characteristics of spinal muscular atrophy with respiratory distress type 1 in the Netherlands [J]. Neuromuscul Disord, 2013, 23(6):461-468.
[5] Messina MF, Messina S, Gaeta M, et al. Infantile spinal muscular atrophy with respiratory distress type I (SMARD 1): an atypical phenotype and review of the literature [J]. Eur J Paediatr Neurol, 2012, 16(1):90-94.
[6] Blaschek A, Glaser D, Kuhn M, et al. Early infantile sensorymotor neuropathy with late onset respiratory distress [J]. Neuromuscul Disord, 2014, 24(3):269-271.
[7] Guenther UP, Varon R, Schlicke M, et al. Clinical and mutational profile in spinal muscular atrophy with respiratory distress (SMARD): defining novel phenotypes through hierarchical cluster analysis [J]. Hum Mutat, 2007, 28(8): 808-815.
[8] Rudnik-Schoneborn S, Forkert R, Hahnen E, et al. Clinical spectrum and diagnostic criteria of infantile spinal muscular atrophy: further delineation on the basis of SMN gene deletion findings [J]. Neuropediatrics, 1996, 27(1):8-15.
[9] Eckart M, Guenther UP, Idkowiak J, et al. The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1) [J]. Pediatrics, 2012, 129(1): e148- e156.
[10] Hamilton MJ, Longman C, O'Hara A, et al. Growing up with spinal muscular atrophy with respiratory distress (SMARD1). [J]. Neuromuscular Disorders, 2015, 25(2): 169-171.
[11] Pierson TM, Tart G, Adams D, et al. Infantile-onset spinal muscular atrophy with respiratory distress-1 diagnosed in a 20-year-old man [J]. Neuromuscul Disord, 2011, 21(5):353-355.
[12] Lim SC, Bowler MW, Lai TF, et al. The Ighmbp2 helicase structure reveals the molecular basis for disease-causing mutations in DMSA1 [J]. Nucleic Acids Res, 2012, 40(21): 11009-11022.
[13] Jedrzejowska M, Madej-Pilarczyk A, Fidzianska A, et al. Severe phenotypes of SMARD1 associated with novel mutations of the IGHMBP2 gene and nuclear degeneration of muscle and Schwann cells [J]. Eur J Paediatr Neurol, 2014, 18(2):183-192.
[14] Kaindl AM, Guenther UP, Rudnik-Schoneborn S, et al. Spinal muscular atrophy with respiratory distress type 1 (SMARD1) [J]. J Child Neurol, 2008, 23(2): 199-204.
[15] Joseph S, Robb SA, Mohammed S, et al. Interfamilial phenotypic heterogeneity in SMARD1 [J]. Neuromuscul Disord, 2009, 19(3):193-195.
[16] Luan X, Huang X, Liu X, et al. Infantile spinal muscular atrophy with respiratory distress type I presenting without respiratory involvement: novel mutations and review of the literature [J]. Brain Dev, 2016, 38(7): 685-689.
[17] Cottenie E, Kochanski A, Jordanova A, et al. Truncating and missense mutations in IGHMBP2 cause Charcot-Marie Tooth disease type 2 [J]. Am J Hum Genet, 2014, 95(5):590601.
[18] 麦嘉卉, 韩春锡. 脊髓性肌萎缩伴呼吸窘迫1型的研究进展[J]. 中华实用儿科临床杂志, 2014, 29(15): 1187-1190.
[19] Markowitz JA, Singh P, Darras BT. Spinal muscular atrophy: a clinical and research update [J]. Pediatr Neurol, 2012, 46(1): 1-12.
[20] Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges [J]. Laheel Neurol, 2012, 11(10): 891-905.
[21] Nizzardo M, Simone C, Rizzo F, et al. Gene therapy rescues disease phenotype in a spinal muscular atrophy with respiratory distress type 1 (SMARD1) mouse model [J]. Sci Adv, 2015, 1(2): e1500078.
Spinal muscular atrophy combined with respiratory distress type I but no respiratory failure: a case report
GUO Li, SUN Longmei, LIU Fang
(Department of Neonatology, People's Liberation Army of Bethune International Peace Hospital, Shijiazhuang 050000, Hebei, China)
ObjectiveTo explore the diagnosis and differential diagnosis of spinal muscular atrophy with respiratory distress type I (SMARD1).MethodThe clinical data, results of gene detection, and follow-up information of a girl diagnosed with SMARD1 were retrospectively analyzed, and related literatures were reviewed.ResultsThe girl was born by cesarean section due to oligohydramnios. After birth, she was transferred to neonatology department for poor feeding and response, and diagnosed with neonatal sepsis, infectious shock, disseminated inravascular coagulation and atypical purulent meningitis. She was discharged after one month of treatment. However, at 2 months old, she presented contracture of ankle joint, abnormal liver function, and myocardial damage. At 6 months old, she had obvious reduced muscular tension and development retardation. At 8 months old, the SMA gene was detected and it was normal. At 9 months old, The panel gene of peripheral neuropathy was detected and found 2 heterozygosis mutations in IGHMBP2 gene, exon8 c.1061-2A>G and exon12 c.1708C>T, which came from her father and mother respectively. Locus of exon12 c.1708C>T has been reported to be associated with the disease, and the other is a shear mutation. The diagnosis of SMARD1 was confirmed by the clinical and gene detection. The girl, 2-year-old now, suffered with recurrent respiratory tract infections, but had no respiratory distress or no respiratory failure yet.ConclusionThe clinical phenotype of SMARD1 is complex and diverse. This case is the first domestic case comfirmed by gene detection.
spinal muscular atrophy with respiratory distress type I; respiratory failure; diaphragmatic paralysis
10.3969/j.issn.1000-3606.2017.03.015
2016-09-19)
(本文编辑:邹 强)
刘芳 电子信箱:liufanglafy@126.com
猜你喜欢
首都食品与医药(2019年4期)2019-10-24
现代电生理学杂志(2016年1期)2016-07-10
中西医结合心脑血管病杂志(2016年20期)2016-03-01
中国卫生标准管理(2015年18期)2016-01-20
中国卫生标准管理(2015年5期)2016-01-14
中国康复理论与实践(2015年7期)2015-05-09
西安交通大学学报(医学版)(2015年2期)2015-02-28
中国卫生标准管理(2015年18期)2015-01-26
中国医药指南(2015年29期)2015-01-24
云南中医学院学报(2014年2期)2014-11-07
