
肢带型肌营养不良症临床特点及诊断与治疗进展
2017-11-23 03:45俞萌王朝霞
中国现代神经疾病杂志 2017年8期
俞萌 王朝霞
·专题综述·
肢带型肌营养不良症临床特点及诊断与治疗进展
俞萌 王朝霞
肢带型肌营养不良症是一组基因突变导致的以近端肌无力为主要表现的遗传性肌病。尽管其临床表现具有共性特征,但不同亚型之间仍存在差异,且遗传缺陷具有异质性,故应结合临床表现、影像学、肌肉病理学和基因检测等综合判断。目前主要采取综合治疗方法。近年来,国内外对此类疾病的诊断与治疗取得新的进展。本文拟就肢带型肌营养不良症各亚型临床特点以及诊断与治疗进展进行概述。
肌营养不良,肢带型; 综述
肢带型肌营养不良症(LGMD)是一组以近端肌(骨盆带肌和肩胛带肌)肌无力为主要表现的遗传性肌病[1⁃2],总患病率为 1.63 ~ 5.00/10 万[3],仅次于抗肌萎缩蛋白病(dystrophinopathy)、面⁃肩⁃肱型肌营养不良症(FSHD)和强直性肌营养不良症1型(DM1型),位居遗传性肌病第4位。肢带型肌营养不良症与基因突变引起的肌细胞蛋白功能缺陷有关,受累蛋白涵盖肌纤维所有结构,如细胞核、胞质、肌小节、肌膜和细胞外基质(ECM)。按照遗传方式可以分为常染色体显性遗传性肢带型肌营养不良症(LGMD1型)和常染色体隐性遗传性肢带型肌营养不良症(LGMD2型)两种类型,每种类型根据致病基因又分为具体临床亚型[4],其中,LGMD1型包括8种亚型(LGMD1A~1H型),LGMD2型包括26种亚型(LGMD2A~2Z型,表1),不同亚型发病年龄、病情严重程度存有差异。近年来,国内外对肢带型肌营养不良症的诊断与治疗取得新的进展,本文拟就肢带型肌营养不良症各亚型临床特点以及诊断与治疗进展进行综述。
一、临床特点
1.LGMD1型 (1)LGMD1A型:系MYOT基因突变所致。发病年龄18~79岁,临床表现为非对称性四肢肌无力和肌萎缩[5],个别患者出现肌肉肥大和肌强直[6],部分伴构音障碍、心肌病和呼吸功能衰竭等[7]。血清肌酸激酶(CK)水平正常或仅轻度升高。影像学显示,腓肠肌内侧头、比目鱼肌和大腿内收肌群受累,半腱肌相对保留[7]。肌肉组织活检可见肌纤维内异常蛋白沉积,伴镶边空泡;抗Myotilin抗体免疫组织化学染色显示,Myotilin蛋白异常聚集;超微结构观察,呈Z线水波纹样改变[6]。(2)LGMD1B型:属LMNA基因相关肌病或层黏连蛋白病范畴。发病年龄2~65岁,临床表现为近端肌无力,下肢重于上肢[8],心脏受累常见。血清肌酸激酶水平正常或仅轻度升高。影像学提示以大腿和小腿后群肌肉为主的脂肪化,腓肠肌内侧头和比目鱼肌受累显著,伴臀大肌、股四头肌、大收肌和腘绳肌受累[9]。肌肉组织活检可见非特异性肌源性改变,应注意的是,部分患儿可见肌内衣炎症性改变,伴镶边空泡;免疫组织化学染色,核纤层蛋白A/C(lamin A/C)表达正常或下降;超微结构观察,细胞核异常伴核周异常染色质丢失或与核膜脱离,以及染色质之间纹理改变[10]。(3)LGMD1C 型:系 CAV3基因突变所致。发病年龄5~81岁,临床呈现骨骼肌高兴奋性特点,表现为显著自发性运动后肌肉痉挛,伴或不伴肌肉疼痛,此外,部分患者还出现特征性叩击诱发的肌肉快速收缩[11]。血清肌酸激酶升高3~30倍。肌肉组织活检正常或者轻微肌病样或肌营养不良样病理改变;免疫组织化学染色,小窝蛋白 3(caveolin⁃3)表达下降[12]。(4)LGMD1D 型:系DNAJB6基因突变所致,分为晚发型和早发严重型。晚发型通常于20~60岁发病,表现为中至重度下肢近端肌无力,而股四头肌和腘绳肌受累程度相对较轻;血清肌酸激酶水平正常或仅轻度升高;影像学提示疾病早期以比目鱼肌、大收肌、半膜肌和股二头肌脂肪化为主,并逐渐累及腓肠肌内侧头、长收肌和股四头肌,而股直肌、腓肠肌外侧头、缝匠肌、股薄肌和小腿前群肌肉相对保留[13]。早发严重型通常于10岁左右发病,早期可以出现肺功能异常、构音障碍和吞咽困难,甚至危及生命;影像学提示腓肠肌外侧头早期受累;肌肉组织活检显示,肌
病样或肌营养不良样病理改变,伴镶边空泡、胞质内包涵体和肌纤维紊乱[14]。(5)LGMD1E 型:系 DES基因突变所致。发病年龄为10~60岁,四肢远端肌无力重于近端,伴早期足下垂,心脏受累是特征性临床表现。血清肌酸激酶水平正常或仅轻度升高。影像学提示臀大肌受累较臀中肌和臀小肌严重,半腱肌、缝匠肌和股薄肌早期受累,股四头肌相对保留,腓骨肌群较胫前肌受累严重,小腿后群肌肉相对保留。肌肉组织活检显示,肌营养不良样病理改变伴无定形物质沉积,或者肌病样病理改变伴胞浆体,继发线粒体功能异常[15];抗结蛋白(desmin)抗体免疫组织化学染色,胞浆体结蛋白阳性[16]。(6)LGMD1F 型:系TNPO3基因突变所致。发病年龄1~31岁,常伴特殊骨骼畸形,如“蜘蛛”样手指和(或)足趾,是该类型的特征性表现;部分可出现吞咽困难、呼吸肌受累,但心脏受累相对罕见。血清肌酸激酶水平正常或仅轻度升高。影像学提示以股外侧肌和小腿三头肌受累为主。肌肉组织活检显示,肌病样病理改变,特征性表现为胞质内嗜碱性物质和酸性磷酸酶阳性的溶酶体空泡[17]。(7)LGMD1G型:系HNRNPDL基因突变所致。发病年龄为15~53岁,病情进展较为缓慢,可独立行走,伴手指和(或)足趾屈曲受限,指和(或)趾骨间关节活动度减少,但手指和(或)足趾背伸力正常,是该类型特征性表现,多合并白内障。血清肌酸激酶水平正常或仅轻度升高。肌肉组织活检显示,肌纤维直径变异增大,偶可见坏死肌纤维伴镶边空泡[18]。(8)LGMD1H型:致病基因尚未明确。发病年龄约50岁,重型患者表现为缓慢进展的四肢近端肌无力;血清肌酸激酶水平轻至中度升高。轻型患者表现为腓肠肌肥大;肌肉组织活检显示,肌纤维直径变异增大,结缔组织增生,伴较多核内移现象[19]。
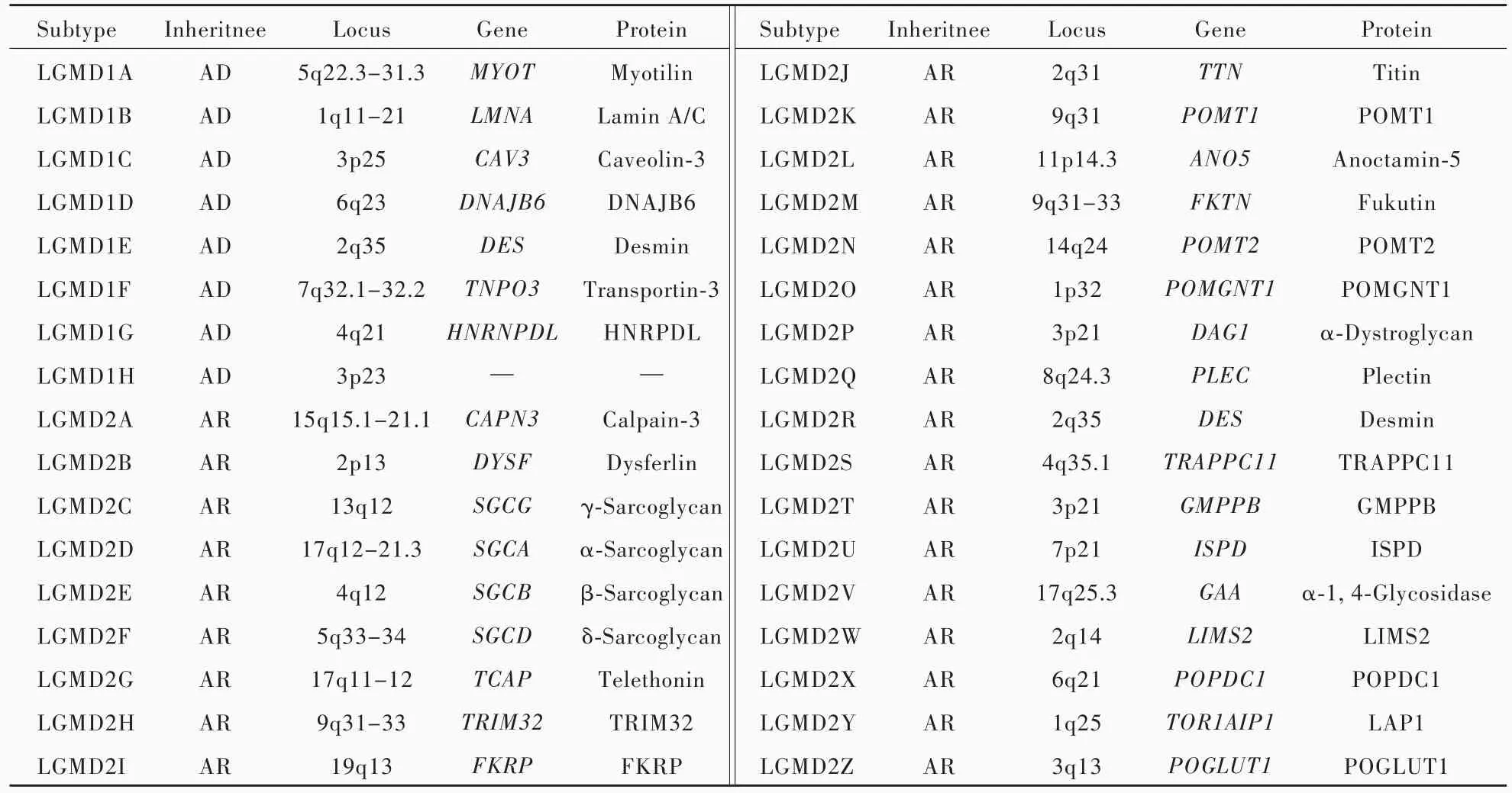
表1 肢带型肌营养不良症分型Table 1. Classification of LGMD
2.LGMD2型 (1)LGMD2A型:致病基因为CAPN3基因。发病年龄2~55岁,高峰发病年龄6~18岁,病情进展缓慢,通常于发病10~30年后丧失行走能力。临床主要表现为跑步困难、翼状肩胛、步态异常和轻度脊柱前凸,心脏受累少见,病程后期可见呼吸肌受累[20]。病程早期血清肌酸激酶水平明显升高,后期逐渐下降。影像学提示疾病早期臀大肌、大腿内收肌群和大腿后群肌肉受累,后期股四头肌受累,而缝匠肌和股薄肌相对保留且常伴肥大,小腿比目鱼肌和腓肠肌内侧头受累,而腓肠肌外侧头和胫骨后肌相对保留[21]。肌肉组织活检显示,肌营养不良样病理改变伴非特异性肌病样改变,分叶状肌纤维是特征性病理改变,且数目随病程进展而增加;免疫印迹法(Western blotting法)检测肌肉Calpain⁃3蛋白表达变化对疾病诊断具有重要意义。此外,亦有符合常染色体显性遗传规律的个案报道[22]。(2)LGMD2B 型:系 DYSF 基因突变所致。通常于青少年晚期或成年早期发病,少数发病前有剧烈运动诱因。肢体近端肌肉受累,下肢重于上肢,随着病情进展,亦累及下肢远端,出现小腿无力和腓肠肌萎缩。上肢症状以肱二头肌无力为主,特征性体征为三角肌凸起,部分萎缩的肱二头肌与选择性萎缩的肩胛带肌特征性表现为“双牛头奖杯征”;大腿肌肉选择性受累,部分表现为股四头肌“钻石凸起征”。面肌、呼吸肌和心肌通常不受累。疾病早期血清肌酸激酶水平显著升高,此后逐渐下降[23]。影像学提示疾病早期大腿肌群中大收肌受累,逐渐累及股外侧肌和半膜肌,最终大腿前群、内侧群和后群肌肉弥漫性受累,而缝匠肌和股薄肌相对保留;小腿肌群中腓肠肌内侧头受累,逐渐累及腓肠肌外侧头和比目鱼肌,而胫骨后肌相对保留;此外,疾病早期还可见骨骼肌水肿改变[24]。肌肉组织活检提示肌营养不良样病理改变伴炎症性改变是其特征性病理改变;免疫组织化学染色,肌膜和胞质Dysferlin蛋白阴性或表达下降[25],可资与炎性肌肉病相鉴别。(3)LGMD2C~2F型:均属肌聚糖蛋白病,致病基因分别为SGCG、SGCA、SGCB、SGCD基因。通常于儿童早期发病,伴翼状肩胛、小腿肥大、巨舌症、关节挛缩和脊柱侧弯,多于青春期丧失行走能力,部分出现呼吸肌无力并需呼吸机辅助通气,心脏受累少见。血清肌酸激酶水平明显升高。影像学提示疾病早期以骨盆带肌尤其是臀肌及大腿后部和前部深在肌群受累为主,而股中间肌相对保留,伴股直肌、缝匠肌和股薄肌肥大,偶见比目鱼肌、腓肠肌和腓骨长肌受累。肌肉组织活检显示,肌营养不良样病理改变;免疫组织化学染色,α、β、γ、δ⁃肌聚糖(sarcoglycan)水平显著降低或缺失[26]。(4)LGMD2G型:系TCAP基因突变所致。发病年龄2~15岁,伴脊柱前凸、翼状肩胛、足下垂和肌肉痉挛,部分可累及心脏。血清肌酸激酶水平轻至中度升高。影像学显示,弥漫性大腿肌群和比目鱼肌、腓肠肌内侧头受累,而胫骨前肌相对保留。肌电图呈现神经源性和肌源性损害。肌肉组织活检可见大量镶边空泡,伴分叶状肌纤维、杆状体、核内移和线 粒 体 类 结 晶 包 涵 体[27]。(5)LGMD2H 型 :系TRIM32基因突变所致。通常于70岁左右发病,部分伴周围神经病变,电生理学表现为神经传导速度(NCV)减慢。血清肌酸激酶水平正常或仅轻度升高。肌肉组织活检显示,大量小空泡出现在以Ⅱ型为主的肌纤维中;超微结构观察显示,小空泡是局部扩张的肌浆内质网,互相融合,伴肌膜变性[28]。(6)LGMD2I型:系FKRP基因突变所致。发病年龄9~23岁,下肢以屈髋肌群和大腿内收肌群受累显著,上肢以肩内收肌群和屈肘肌群显著,伴腓肠肌肥大、肌肉疼痛和疲劳感,多合并扩张型心肌病,甚至发生致死性心功能衰竭,呼吸功能障碍较为常见,表现为限制性通气功能障碍[29]。血清肌酸激酶水平升高10倍以上,伴阵发性肌红蛋白尿。影像学显示,腰大肌、臀大肌和大腿内收肌群受累,而大腿前群肌肉相对保留,缝匠肌和股薄肌受累较晚或不受累。肌肉组织活检显示,肌营养不良样病理改变;免疫组织化学染色显示,α⁃肌营养不良蛋白聚糖(α⁃dystroglycan)水平下降[30]。(7)LGMD2J型:系TTN基因突变所致。通常于30岁前发病,临床主要表现为重度近端肌无力,伴轻至中度远端肌无力,部分可伴心律失常,呼吸肌受累相对少见。血清肌酸激酶水平轻度升高。影像学显示,小腿肌群脂肪化,大腿前群或前群和后群肌肉同时受累,而外侧肌群相对保留。肌肉组织活检显示,肌营养不良样病理改变[31]。(8)LGMD2K 型:系 POMT1 基因突变所致。通常于婴儿期或儿童早期发病,伴小头畸形和智力缺陷。血清肌酸激酶水平升高10倍以上。头部MRI检查正常或仅轻微白质改变。肌肉组织活检显示,肌纤维直径变异增大。免疫组织化学染色显示,α⁃肌营养不良蛋白聚糖表达水平下降[32]。(9)LGMD2L型:系ANO5基因突变所致。发病年龄20~57岁,部分伴腓肠肌肥大,多数表现为股四头肌和肱二头肌不对称性肌萎缩,心脏和肺受累罕见,晚发型患者肌无力症状轻微。血清肌酸激酶水平升高8~20倍。影像学提示以臀小肌以及大腿和小腿后群肌肉受累为主,而小腿前群肌肉、缝匠肌和股薄肌相对保留。肌肉组织活检显示,肌病样或肌营养不良样病理改变[33]。(10)LGMD2M 型:系FKTN基因突变所致。通常于4个月至14岁发病,认知功能正常,常累及心脏。血清肌酸激酶水平显著升高。肌肉组织活检显示,肌营养不良样病理改变伴炎症性改变;免疫组织化学染色,α⁃肌营养不良蛋白聚糖缺失[34]。(11)LGMD2N 型:系 POMT2 基因突变所致。通常于婴儿期或儿童早期发病,临床主要表现为运动发育迟滞并逐渐出现肢带肌无力。血清肌酸激酶水平显著升高。肌肉组织活检显示,肌营养不良样病理改变伴炎性细胞浸润;免疫组织化学染色,α⁃肌营养不良蛋白聚糖水平下降[35]。(12)LGMD2O型:系POMGNT1基因突变所致。儿童早期至青年期发病,认知功能和心肺功能正常。血清肌酸激酶水平显著升高。肌肉组织活检可见肌营养不良样病理改变;免疫组织化学染色,α⁃肌营养不良蛋白聚糖水平下降[36]。(13)LGMD2P 型:系DAG1基因突变所致。目前仅见1例个案报道,3岁发病,伴小头畸形、脊柱前凸、踝关节挛缩和显著智力发育迟滞。血清肌酸激酶水平升高10倍以上。免疫组织化学染色,选择性α⁃肌营养不良蛋白聚糖水平显著下降[37]。(14)LGMD2Q 型:系 PLEC 基因突变所致。通常于2~3岁发病,约20岁丧失行走能力,心功能正常。该基因突变可以引起大疱性表皮松解症伴肌营养不良症,同时可伴复视、眼外肌瘫痪、运动发育迟滞、面肌无力、龋齿、指甲营养不良、瘢痕性秃发、尿道狭窄、幽门闭锁、食管狭窄、呼吸阻塞和心肌病等。血清肌酸激酶水平显著升高。肌肉组织活检可见肌原纤维肌病样病理改变[29]。(15)LGMD2R型:系DES基因突变所致。青少年期发病,伴关节挛缩、翼状肩胛、脊柱侧弯,而心肌病少见。血清肌酸激酶水平正常。肌肉组织活检显示肌营养不良样病理改变[38]。(16)LGMD2S 型:系TRAPPC11基因突变所致。通常于学龄早期发病,伴肌肉疼痛和肌肉痉挛,部分伴骨骼异常,包括髋关节发育不良和脊柱侧弯,亦可伴白内障、集合性斜视,部分患者可出现智力发育迟滞以及舞蹈样动作、手足徐动、震颤和肌张力障碍性姿势,亦可出现癫发作,而心肺功能正常。血清肌酸激酶轻至中度升高。肌肉组织活检显示肌病样病理改变[39]。(17)LGMD2T型:系GMPPB基因突变所致。通常于出生后至成年期发病,部分伴阵发性横纹肌溶解症、轻度智力发育迟滞、癫发作、小头畸形、白内障、眼震等,心肺功能正常。血清肌酸激酶水平中至重度升高。肌肉组织活检,肌营养不良样病理改变伴核内移和炎性细胞浸润[40]。(18)LGMD2U 型:系ISPD基因突变所致。通常于儿童早期发病,多伴广泛性肌肉肥大,部分伴舌体肥大,并出现翼状肩胛、脊柱侧弯、运动诱发性肌肉痉挛等,心功能轻度下降,部分可出现夜间低通气、呼吸功能障碍等。血清肌酸激酶水平轻至中度升高。影像学提示下肢肌肉脂肪化,缝匠肌、股薄肌和胫骨前后肌相对保留。肌肉组织活检,肌营养不良样病理改变[41]。(19)LGMD2V型:系GAA基因突变所致。青年期发病,伴糖原沉积病Ⅱ型的特点,即与四肢肌肉受累程度不成比例的中轴肌和呼吸肌受累以及特征性病理改变等[42]。(20)LGMD2W 型:系 LIMS2 基因突变所致。目前仅见1个家系报道,于5岁发病,伴小腿肥大和巨舌症,呈现“三角”形舌外观,合并扩张型心肌病;血清肌酸激酶水平中至重度升高;影像学显示,双侧大腿肌肉对称性弥漫性重度萎缩和脂肪化;肌肉组织活检,肌营养不良样病理改变[43]。(21)LGMD2X型:系POPDC1基因突变所致。目前仅见1个家系报道,青中年发病,伴心脏受累,以传导阻滞为主;血清肌酸激酶水平中至重度升高,尤以运动后升高显著;肌肉组织活检显示,肌营养不良样病理改变伴核内移增加;超微结构观察,肌膜连续性中断伴膜下镶边空泡[44]。(22)LGMD2Y 型:系TOR1AIP1基因突变所致。目前仅见1个家系报道,婴儿期出现运动发育迟缓,伴眼睑下垂、脊柱前凸、关节挛缩,此后出现心脏受累,表现为扩张型心肌病,肺功能正常;血清肌酸激酶正常或仅轻度升高;影像学显示,下肢广泛性肌萎缩伴脂肪化,而臀大肌和大腿内收肌群相对保留。肌肉组织活检显示肌营养不良样病理改变;超微结构观察,肌细胞核膜异常[45]。(23)LGMD2Z 型:系 POGLUT1 基因突变所致。目前仅见1个家系报道,20岁后发病,病情进展缓慢,翼状肩胛;血清肌酸激酶正常或仅轻度升高;影像学显示,疾病早期以大腿内侧肌群受累为主,逐渐出现外侧肌群受累;肌肉组织活检显示,轻微肌病样或肌营养不良样病理改变[46]。
二、主要辅助检查
对于疑似肌肉病的患者,血清肌酸激酶检测和肌电图检查均可协助定位诊断,主要包括:(1)骨骼肌影像学检查。骨骼肌CT和MRI均为无创伤性检查方法,可以评价肌肉水肿、脂肪浸润、肌萎缩和肌肉肥大程度,以及判断肌肉受累模式,对诊断和分型具有指导意义。一项采用骨骼肌CT评价118例肢带型肌营养不良症患者的临床研究显示,其诊断分型灵敏度为40%、特异度为58%[47],提示骨骼肌CT可以作为辅助诊断工具。目前,越来越多针对肢带型肌营养不良症各亚型骨骼肌影像学改变规律的研究逐渐揭示不同亚型肌群受累特点,为肢带型肌营养不良症的诊断提供更多依据[24]。(2)肌肉组织活检术。肢带型肌营养不良症肌肉病理改变主要呈现肌营养不良样病理改变特点,即肌纤维直径变异增大,出现肌纤维肥大、萎缩、坏死、再生和结缔组织增生,并伴肌纤维分裂、镶边空泡、分叶状肌纤维等,部分亚型还可表现为肌病样病理改变。随着病程进展,肌纤维逐渐被脂肪和结缔组织替代。由于其发病机制涉及肌细胞蛋白缺陷,采用特异性抗体行免疫组织化学染色或免疫印迹法测定蛋白质含量对特定亚型的诊断具有重要价值[48]。(3)基因检测。通过基因检测发现致病性突变,并通过生物学信息分析,最终实现分子生物学诊断是肢带型肌营养不良症的最终诊断和分型依据。国外指南推荐Sanger测序技术对疑似亚型的致病基因进行检测,并结合家系保守性和致病性分析,最终明确诊断[49]。然而,传统基因检测阳性检出率较低,且时间和经济成本较高。随着二代基因测序技术的发展,越来越多的研究采用目标区域捕获测序、全外显子组测序(WES)等方法,其阳性检出率达16%~65%,提示二代基因测序技术在肢带型肌营养不良症的诊断中具有广阔应用前景[50]。但是由于肢带型肌营养不良症与其他肌肉病以及肢带型肌营养不良症各亚型之间存在临床表型重叠,正确解读基因检测结果以及必要时结合肌肉病理改变,对于保证诊断准确性尤为重要。
三、诊断与鉴别诊断
肢带型肌营养不良症各亚型临床表型的重叠性和异质性,给其精确分型带来极大挑战,应综合肌无力分布、遗传方式、特异性临床表现和肌肉病理改变,进行针对性基因检测,最终实现分子生物学诊断。肢带型肌营养不良症主要出现近端肩胛带肌和骨盆带肌受累倾向,亦可累及远端肌肉。除骨骼肌受累外,还可以合并全身多系统表现,主要包括:(1)骨骼系统,关节挛缩、脊柱强直、脊柱前凸、脊柱侧弯、高弓足、高腭弓等。(2)中枢神经系统,认知功能障碍、颅骨畸形、发育里程碑迟滞等。(3)心肺系统,心律失常、心肌病、心源性卒中、呼吸困难、夜间呼吸暂停、打鼾等。(4)皮肤,皮肤改变等。其中,部分特征性临床表现对特定亚型具有较高的诊断敏感性和特异性,例如,显著的自发性、运动后肌肉痉挛和叩击诱发的肌肉快速收缩高度提示LGMD1C型[11]。结合发病特点、肌无力分布、骨骼肌和各系统表现、血清肌酸激酶水平,可以有效提示诊断和分型,指导有针对性的基因检测。随着基因检测技术的发展,二代基因测序技术在遗传性肌病,尤其是肢带型肌营养不良症的诊断中越来越发挥重要作用。但是对于检测结果的解读和致病性突变的分析还应结合临床表现和病理改变等,最终实现分子生物学诊断。
肢带型肌营养不良症的鉴别诊断主要有炎性肌病、其他遗传性肌营养不良症(如抗肌萎缩蛋白病、面⁃肩⁃肱型肌营养不良症、Emery⁃Dreifuss型肌营养不良症、强直性肌营养不良症)、先天性肌病、代谢性肌病、重症肌无力(MG)和先天性肌无力综合征、脊髓性肌萎缩症(SMA)、内分泌性和中毒性肌病等,应根据肌无力特点、肌肉外受累表现、电生理学、免疫学、肌肉影像学、肌肉病理学、基因检测和药物治疗反应等进行鉴别诊断。
四、治疗
目前对于肢带型肌营养不良症尚缺乏有效治疗方法,康复训练可以延缓疾病进展、改善临床症状,针对合并症的治疗对改善预后具有重要作用。目前,基因治疗已在动物实验中取得一定疗效,带来新的曙光。
1.临床康复管理 其目的是尽量长时间地维持肢体活动和功能,使生活质量最优化,并预防和管理并发症。应联合多学科(如物理疗法、职业治疗、呼吸功能训练、言语功能训练、吞咽功能训练,以及心脏病学、呼吸病学、骨科学和遗传学)制定个体化治疗方案。
2.力量训练和有氧运动训练 研究显示,力量训练和有氧运动训练可以使肢带型肌营养不良症患者受益。现有研究显示其相对安全,无明显不良反应[51]。但是由于肌肉退行性变,高强度训练可能诱发肌肉损伤、肌红蛋白尿和肌无力。因此,建议患者进行有氧运动训练结合次最大强度力量训练,同时,温和、低强度的有氧运动训练(如游泳,静态骑脚踏车)可以改善心功能、提高肌肉效率、减轻疲劳感。
3.药物治疗及其他 目前对基因转导、肌母细胞移植、抗肌肉生长抑制素(MSTN)抗体和生长激素等的长期有效性和安全性尚缺乏证据。腺相关病毒(AAV)载体已在部分动物模型和小型临床试验中初步观察到肯定疗效且无严重不良反应[52]。采用成簇的规律间隔的短回文重复序列(CRISPR)及其相关蛋白Cas9系统进行基因编辑亦在部分动物模型中获得成功[53]。此外,通过诱导型多能干细胞(iPSCs)和基因编辑技术纠正致病性突变并分化、移植,已在动物模型中观察到临床表型改善[54]。
综上所述,随着遗传学、分子生物学、生物化学等的发展,肢带型肌营养不良症疾病谱系将进一步扩展,我们对疾病发病机制、临床表型的认识也将进一步加深。新兴治疗方法有望由实验室引入临床,为此类疾病的诊断与治疗带来新的希望。
[1]Walton JN,Nattrass FJ.On the classification,natural history and treatment of the myopathies.Brain,1954,77:169⁃231.
[2]Bushby KM.Diagnostic criteria for the limb⁃girdle muscular dystrophies:report of the ENMC consortium on limb⁃girdle dystrophies.Neuromuscul Disord,1995,5:71⁃74.
[3]Mah JK,Korngut L,Fiest KM,Dykeman J,Day LJ,Pringsheim T,Jette N.A systematic review and Meta⁃analysis on the epidemiology of the muscular dystrophies.Can J Neurol Sci,2016,43:163⁃177.
[4]Waite A,Tinsley CL,Locke M,Blake DJ.The neurobiology of the dystrophin⁃associated glycoprotein complex.Ann Med,2009,41:344⁃359.
[5]Rudolf G,Suominen T,Penttila S,Hackman P,Evila A,Lannes B,Echaniz⁃Laguna A,Bierry G,TranchantC,Udd B.Homozygosity of the dominant myotilin c.179C>T(p.Ser60Phe)mutation causes a more severe and proximal muscular dystrophy.J Neuromuscul Dis,2016,3:275⁃281.
[6]Olivé M,Odgerel Z,Martínez A,Poza JJ,Bragado FG,Zabalza RJ,Jericó I,Gonzalez⁃Mera L,Shatunov A,Lee HS,Armstrong J,Maraví E,Arroyo MR,Pascual⁃Calvet J,Navarro C,Paradas C,Huerta M,Marquez F,Rivas EG,Pou A,Ferrer I,Goldfarb LG.Clinical and myopathological evaluation of early⁃and late⁃onset subtypes of myofibrillar myopathy.Neuromuscul Disord,2011,21:533⁃542.
[7]Reilich P,Krause S,Schramm N,Klutzny U,BulstS,Zehetmayer B, Schneiderat P, Walter MC, Schoser B,Lochmuller H.A novel mutation in the myotilin gene(MYOT)causes a severe form of limb girdle muscular dystrophy 1A(LGMD1A).J Neurol,2011,258:1437⁃1444.
[8]Furuta M,Sumi⁃Akamaru H,Takahashi MP,Hayashi YK,Nishino I,Mochizuki H.An elderly⁃onset limb girdle muscular dystrophytype1B (LGMD1B)with pseudo⁃hypertrophyof paraspinal muscles.Neuromuscul Disord,2016,26:593⁃597.
[9]Komaki H,Hayashi YK,Tsuburaya R,Sugie K,Kato M,Nagai T,ImatakaG,Suzuki S,Saitoh S,AsahinaN,HonkeK,Higuchi Y,Sakuma H,Saito Y,Nakagawa E,Sugai K,Sasaki M,Nonaka I,Nishino I.Inflammatory changes in infantile⁃onset LMNA⁃associated myopathy.Neuromuscul Disord,2011,21:563⁃568.
[10]Sabatelli P,Lattanzi G,Ognibene A,Columbaro M,Capanni C,Merlini L,Maraldi NM,Squarzoni S.Nuclear alterations in autosomal⁃dominantEmery⁃Dreifuss musculardystrophy.Muscle Nerve,2001,24:826⁃829.
[11]ScalcoRS,GardinerAR,PitceathlyRD,Hilton⁃JonesD,Schapira AH,Turner C,Parton M,Desikan M,Barresi R,Marsh J,Manzur AY,Childs AM,Feng L,Murphy E,Lamont PJ,Ravenscroft G,Wallefeld W,Davis MR,Laing NG,Holton JL,Fialho D,Bushby K,Hanna MG,Phadke R,Jungbluth H,Houlden H,Quinlivan R.CAV3 mutations causing exercise intolerance, myalgia and rhabdomyolysis: expanding the phenotypic spectrum of caveolinopathies.Neuromuscul Disord,2016,26:504⁃510.
[12]González⁃Pérez P,Gallano P,Gonzalez⁃Quereda L,Rivas⁃Infante E,Teijeira S,Navarro C,Bautista⁃Lorite J.Phenotypic variability in a Spanish family with a Caveolin⁃3 mutation.J Neurol Sci,2009,276:95⁃98.
[13]Sandell SM,Mahjneh I,Palmio J,Tasca G,Ricci E,Udd BA.'Pathognomonic'muscle imaging findings in DNAJB6 mutated LGMD1D.Eur J Neurol,2013,20:1553⁃1559.
[14]Ruggieri A,Brancati F,Zanotti S,Maggi L,Pasanisi MB,Saredi S,Terracciano C,Antozzi C,D Apice MR,Sangiuolo F,Novelli G,Marshall CR,Scherer SW,Morandi L,Federici L,Massa R,Mora M,Minassian BA.Complete loss of the DNAJB6 G/F domain and novel missense mutations cause distal⁃onset DNAJB6 myopathy.Acta Neuropathol Commun,2015,3:44.
[15]Winter L,Wittig I,Peeva V,Eggers B,Heidler J,Chevessier F,Kley RA,Barkovits K,Strecker V,Berwanger C,Herrmann H,Marcus K,Kornblum C,Kunz WS,Schroder R,Clemen CS.Mutant desmin substantially perturbs mitochondrial morphology,function and maintenance in skeletalmuscle tissue.Acta Neuropathol,2016,132:453⁃473.
[16]Bergman JE,Veenstra⁃KnolHE,van Essen AJ,van Ravenswaaij CM,den Dunnen WF,van den Wijngaard A,van Tintelen JP.Two related Dutch families with a clinically variable presentation of cardioskeletal myopathy caused by a novel S13F mutation in the desmin gene.Eur J Med Genet,2007,50:355⁃366.
[17]Cenacchi G,Peterle E,Fanin M,Papa V,Salaroli R,Angelini C.Ultrastructural changes in LGMD1F.Neuropathology,2013,33:276⁃280.
[18]Vieira NM,Naslavsky MS,Licinio L,Kok F,Schlesinger D,VainzofM,SanchezN,KitajimaJP,GalL,CavacanaN,Serafini PR,Chuartzman S,Vasquez C,Mimbacas A,Nigro V,Pavanello RC,Schuldiner M,Kunkel LM,Zatz M.A defect in the RNA ⁃processing protein HNRPDL causes limb⁃girdle muscular dystrophy 1G(LGMD1G).Hum Mol Genet,2014,3:4103⁃4110.
[19]Bisceglia L, Zoccolella S, Torraco A, Piemontese MR,Dell'Aglio R,Amati A,De Bonis P,Artuso L,Copetti M,Santorelli FM,Serlenga L,Zelante L,Bertini E,Petruzzella V.A new locus on 3p23-p25 for an autosomal⁃dominant limb ⁃girdle muscular dystrophy,LGMD1H.Eur J Hum Genet,2010,18:636⁃641.
[20]Mori⁃Yoshimura M,Segawa K,Minami N,Oya Y,Komaki H,Nonaka I,Nishino I,Murata M.Cardiopulmonary dysfunction in patients with limb⁃girdle muscular dystrophy 2A.Muscle Nerve,2017,55:465⁃469.
[21]Degardin A,Morillon D,Lacour A,Cotten A,Vermersch P,Stojkovic T.Morphologic imaging in muscular dystrophies and inflammatory myopathies.Skeletal Radiol,2010,39:1219⁃1227.
[22]Vissing J,Barresi R,Witting N,Van Ghelue M,Gammelgaard L,Bindoff LA,Straub V,Lochmuller H,Hudson J,Wahl CM,Arnardottir S,Dahlbom K,Jonsrud C,Duno M.A heterozygous 21 bp deletion in CAPN3 causes dominantly inherited limb girdle muscular dystrophy.Brain,2016,139:2154⁃2163.
[23]Jin SQ,Yu M,Zhang W,Lyu H,Yuan Y,Wang ZX.Dysferlin gene mutation spectrum in a large cohort of chinese patients with dysferlinopathy.Chin Med J(Engl),2016,129:2287⁃2293.
[24]Jin S,Du J,Wang Z,Zhang W,Lv H,Meng L,Xiao J,Yuan Y.Heterogeneous characteristics of MRI changes of thigh muscles in patients with dysferlinopathy.Muscle Nerve,2016,54:1072⁃1079.
[25]Nilsson MI,Laureano ML,Saeed M,Tarnopolsky MA.Dysferlin aggregation in limb⁃girdle muscular dystrophy type 2B/Miyoshi Myopathy necessitates mutational screen for diagnosis.Muscle Nerve,2013,47:740⁃747.
[26]Dınız G,Hazan F,Yildirim HT,Unalp A,Polat M,Serdaroglu G,Güzel O,Bag O,Seçıl Y,Ozgönül F,Türe S,Akhan G,Tükün A.Histopathological and genetic features of patients with limb girdle muscular dystrophy type 2C.Turk Patoloji Derg,2014,30:111⁃117.
[27]Paim JF,Cotta A,Vargas AP,Navarro MM,Valicek J,Carvalho E,da⁃Cunha AL Jr,Plentz E,Braz SV,Takata RI,Almeida CF,Vainzof M. Muscle phenotypic variability in limb girdle muscular dystrophy 2G.J Mol Neurosci,2013,50:339⁃344.
[28]Borg K,Stucka R,Locke M,Melin E,Ahlberg G,Klutzny U,Hagen Mv,Huebner A,Lochmuller H,Wrogemann K,Thornell LE,Blake DJ,Schoser B.Intragenic deletion of TRIM32 in compound heterozygotes with sarcotubular myopathy/LGMD2H.Hum Mutat,2009,30:E831⁃844.
[29]FattahiZ,KahriziK,NafissiS,Fadaee M,Abedin SS,Kariminejad A,Akbari MR,Najmabadi H.Report of a patient with limb⁃girdle muscular dystrophy, ptosis and ophthalmoparesis caused by plectinopathy.Arch Iran Med,2015,18:60⁃64.
[30]Fu X,Yang H,Wei C,Jiao H,Wang S,Yang Y,Han C,Wu X,Xiong H.FKRP mutations,including a founder mutation,cause phenotype variability in Chinese patients with dystroglycanopathies.J Hum Genet,2016,61:1013⁃1020.
[31]Pénisson ⁃Besnier I,Hackman P,Suominen T,Sarparanta J,Huovinen S,Richard⁃Crémieux I,Udd B.Myopathies caused by homozygous titin mutations:limb⁃girdle muscular dystrophy 2J and variations of phenotype.J Neurol Neurosurg Psychiatry,2010,81:1200⁃1202.
[32]Lommel M,Cirak S,Willer T,Hermann R,Uyanik G,van Bokhoven H,Korner C,Voit T,Baric I,Hehr U,Strahl S.Correlation of enzyme activity and clinical phenotype in POMT1⁃associated dystroglycanopathies.Neurology,2010,74:157⁃164.
[33]SchneiderI,StoltenburgG,DeschauerM,WinterhollerM,Hanisch F.Limb girdle muscular dystrophy type 2L presenting as necrotizing myopathy.Acta Myol,2014,33:19⁃21.
[34]Godfrey C,Escolar D,Brockington M,Clement EM,Mein R,Jimenez⁃Mallebrera C,Torelli S,Feng L,Brown SC,Sewry CA,Rutherford M,Shapira Y,Abbs S,Muntoni F.Fukutin gene mutations in steroid⁃responsive limb girdle muscular dystrophy.Ann Neurol,2006,60:603⁃610.
[35]Biancheri R,Falace A,Tessa A,Pedemonte M,Scapolan S,Cassandrini D,Aiello C,Rossi A,Broda P,Zara F,Santorelli FM,Minetti C,Bruno C.POMT2 gene mutation in limb⁃girdle muscular dystrophy with inflammatory changes. Biochem Biophys Res Commun,2007,363:1033⁃1037.
[36]Clement EM,Godfrey C,Tan J,Brockington M,Torelli S,Feng L,Brown SC,Jimenez⁃Mallebrera C,Sewry CA,Longman C,Mein R,Abbs S,Vajsar J,Schachter H,Muntoni F.Mild POMGnT1 mutations underlie a novel limb⁃girdle muscular dystrophy variant.Arch Neurol,2008,65:137⁃141.
[37]Hara Y,Balci⁃Hayta B,Yoshida⁃Moriguchi T,Kanagawa M,Beltrán ⁃Valero de Bernabé D,Gündeşli H,Willer T,Satz JS,Crawford RW,Burden SJ,Kunz S,Oldstone MB,Accardi A,Talim B,Muntoni F,Topaloglu H,Dinçer P,Campbell KP.A dystroglycan mutation associated with limb⁃girdle muscular dystrophy.N Engl J Med,2011,364:939⁃946.
[38]Cetin N,Balci⁃Hayta B,Gundesli H,Korkusuz P,Purali N,Talim B,Tan E,Selcen D,Erdem⁃Ozdamar S,Dincer P.A novel desmin mutation leading to autosomal recessive limb⁃girdle muscular dystrophy:distinct histopathological outcomes compared with desminopathies.J Med Genet,2013,50:437⁃443.
[39]Bögershausen N,Shahrzad N,Chong JX,von Kleist⁃Retzow JC,Stanga D,Li Y,Bernier FP,Loucks CM,Wirth R,Puffenberger EG,Hegele RA,Schreml J,Lapointe G,Keupp K,Brett CL,Anderson R,Hahn A,Innes AM,Suchowersky O,Mets MB,Nürnberg G,McLeod DR,Thiele H,Waggoner D,Altmüller J,BoycottKM,SchoserB,Nürnberg P,OberC,HellerR,Parboosingh JS,Wollnik B,Sacher M,Lamont RE.Recessive TRAPPC11 mutations cause a disease spectrum of limb girdle muscular dystrophy and myopathy with movement disorder and intellectual disability.Am J Hum Genet,2013,93:181⁃190.
[40]Oestergaard ST,Stojkovic T,Dahlqvist JR,Bouchet⁃Seraphin C,Nectoux J,Leturcq F,Cossee M,Sole G,Thomsen C,Krag TO,Vissing J.Muscle involvementin limb⁃girdle muscular dystrophy with GMPPB deficiency(LGMD2T).Neurol Genet,2016,2:E112.
[41]Cirak S,Foley AR,Herrmann R,Willer T,Yau S,Stevens E,Torelli S,BroddL,KamyninaA,VondracekP,RoperH,Longman C,Korinthenberg R,Marrosu G,Nurnberg P;UK 10K Consortium;Michele DE,Plagnol V,Hurles M,Moore SA,SewryCA,CampbellKP,VoitT,MuntoniF.ISPD gene mutations are a common cause of congenital and limb⁃girdle muscular dystrophies.Brain,2013,136:269⁃281.
[42]Preisler N,Lukacs Z,Vinge L,Madsen KL,Husu E,Hansen RS,Duno M,Andersen H,Laub M,Vissing J.Late⁃onset Pompe disease is prevalent in unclassified limb⁃girdle muscular dystrophies.Mol Genet Metab,2013,110:287⁃289.
[43]Chardon JW,Smith AC,Woulfe J,Pena E,Rakhra K,Dennie C,Beaulieu C,Huang L,SchwartzentruberJ,HawkinsC,Harms MB,Dojeiji S,Zhang M;Forge Canada Consortium;Majewski J,Bulman DE,Boycott KM,Dyment DA.LIMS2 mutationsare associated with a novelmusculardystrophy,severe cardiomyopathy and triangulartongues.Clin Genet,2015,88:558⁃564.
[44]Schindler RF,Scotton C,Zhang J,Passarelli C,Ortiz⁃Bonnin B,Simrick S,Schwerte T,Poon KL,Fang M,Rinne S,Froese A,Nikolaev VO,Grunert C,Muller T,Tasca G,Sarathchandra P,Drago F,Dallapiccola B,Rapezzi C,Arbustini E,Di Raimo FR,Neri M,Selvatici R,Gualandi F,Fattori F,Pietrangelo A,Li W,Jiang H,Xu X,BertiniE,Decher N,Wang J,Brand T,Ferlini A.POPDC1(S201F)causes muscular dystrophy and arrhythmia by affecting protein trafficking.J Clin Invest,2016,126:239⁃253.
[45]Ghaoui R,Benavides T,Lek M,Waddell LB,Kaur S,North KN,MacArthur DG,Clarke NF,Cooper ST.TOR1AIP1 as a cause of cardiac failure and recessive limb⁃girdle muscular dystrophy.Neuromuscul Disord,2016,26:500⁃503.
[46]Servian⁃Morilla E,Takeuchi H,Lee TV,Clarimon J,Mavillard F,Area⁃Gomez E,Rivas E,Nieto⁃Gonzalez JL,Rivero MC,Cabrera⁃Serrano M,Gomez⁃Sanchez L,Martinez⁃Lopez JA,Estrada B,Marquez C,Morgado Y,Suarez⁃Calvet X,Pita G,BigotA,Gallardo E,Fernandez⁃Chacon R,Hirano M,HaltiwangerRS,Jafar⁃NejadH,ParadasC.A POGLUT1 mutation causesamusculardystrophy with reduced Notch signaling and satellite cell loss.EMBO Mol Med,2016,8:1289⁃1309.
[47]ten Dam L,van der Kooi AJ,van Wattingen M,de Haan RJ,de Visser M.Reliability and accuracy of skeletal muscle imaging in limb⁃girdle muscular dystrophies.Neurology,2012,79:1716⁃1723.
[48]Bastian A,Mageriu V,Micu G,Manole E.The growing family of limb⁃girdle muscular dystrophies:old and newly identified members.Rom J Intern Med,2015,53:13⁃24.
[49]Narayanaswami P,Weiss M,Selcen D,David W,Raynor E,Carter G,Wicklund M,Barohn RJ,Ensrud E,Griggs RC,Gronseth G,Amato AA;Guideline Development Subcommittee of the American Academy of Neurology;Practice Issues Review Panelofthe American Association ofNeuromuscular &Electrodiagnostic Medicine.Evidence⁃based guideline summary:diagnosis and treatment of limb⁃girdle and distal dystrophies.Reportofthe guideline developmentsubcommittee ofthe American Academy of Neurology and the practice issues review panelofthe American Association ofNeuromuscular &Electrodiagnostic Medicine.Neurology,2014,83:1453⁃1463.
[50]Yu M,Zheng Y,Jin S,Gang Q,Wang Q,Yu P,Lv H,Zhang W,Yuan Y,Wang Z.Mutational spectrum of Chinese LGMD patients by targeted next⁃generation sequencing.PLoS One,2017,12:E0175343.
[51]Siciliano G,Simoncini C,Giannotti S,Zampa V,Angelini C,Ricci G.Muscle exercise in limb girdle muscular dystrophies:pitfall and advantages.Acta Myol,2015,34:3⁃8.
[52]Herson S,Hentati F,Rigolet A,Behin A,Romero NB,Leturcq F,Laforet P,Maisonobe T,Amouri R,Haddad H,Audit M,Montus M,Masurier C,Gjata B,Georger C,Cherai M,Carlier P,Hogrel JY,Herson A,Allenbach Y,Lemoine FM,Klatzmann D,Sweeney HL,Mulligan RC,Eymard B,Caizergues D,Voit T,Benveniste O.A phaseⅠtrial of adeno⁃associated virus serotype 1⁃gamma⁃sarcoglycan gene therapy for limb girdle muscular dystrophy type 2C.Brain,2012,135:483⁃492.
[53]Bengtsson NE,Seto JT,Hall JK,Chamberlain JS,Odom GL.Progress and prospects of gene therapy clinical trials for the muscular dystrophies.Hum Mol Genet,2015,25:9⁃17.
[54]Turan S,Farruggio AP,Srifa W,Day JW,Calos MP.Precise correction of disease mutations in induced pluripotent stem cells derived from patients with limb girdle muscular dystrophy.Mol Ther,2016,24:685⁃696.
Advances in diagnosis and treatment of limb⁃girdle muscular dystrophy
YU Meng,WANG Zhao⁃xia
Department of Neurology,Peking University First Hospital,Beijing 100034,China
Corrseponding author:WANG Zhao⁃xia(Email:drwangzx@163.com)
Limb ⁃girdle muscular dystrophy(LGMD)is a group of disorders caused by gene mutations,with proximal muscle weakness as their main manifestation.Although various subtypes of LGMD share the common feature,heterogenity exist both in clinical phenotype and genetic defects.The diagnosis should be based on the combination of the clinical symptoms,muscle imaging findings,myo⁃pathological changes and genetic testing. Multi⁃discipline management is currently for patients. There has been progress in the diagnosis and treatment of LGMD worldwide in recent years.This review will summarize the advances in the LGMD diagnosis,treatment as well as clinical features of different subtypes of LGMD in order to improve the understanding of LGMD.
Muscular dystrophies,limb⁃girdle; Review
This study was supported by the National Natural Science Foundation of China(No.81571219)and Beijing Municipal Science and Technology Basic Scientific Research Plan Programme (No.Z151100003915126).
10.3969/j.issn.1672⁃6731.2017.08.005
国家自然科学基金资助项目(项目编号:81571219);北京市科学技术委员会专项基础科研项目(项目编号:Z151100003915126)
100034北京大学第一医院神经内科
王朝霞(Email:drwangzx@163.com)
2017⁃05⁃31)
下期内容预告本刊2017年第9和10期报道专题为睡眠障碍,重点内容包括:提高对不宁腿综合征规范诊断与治疗的认识;《中国成人失眠诊断与治疗指南》解读;中国不宁腿综合征研究进展:中国学者海外报道;发作性睡病研究进展;自身免疫性脑炎与睡眠障碍;快速眼动睡眠期行为障碍与神经变性病发病机制研究进展;不同脑区对睡眠及相关运动障碍调控机制研究进展;快速眼动睡眠期行为障碍黑质异常功能连接研究;青少年失眠和睡眠质量及相关因素分析;多导睡眠图监测在肌萎缩侧索硬化症患者睡眠障碍和睡眠呼吸障碍事件中的应用价值;早期帕金森病患者快速眼动睡眠期行为障碍研究;缺血性脑血管病合并不宁腿综合征相关因素分析;罕见成人睡眠相关节律性运动障碍一例
猜你喜欢
冰雪运动(2021年1期)2021-07-28
中华养生保健(2021年18期)2021-02-13
安徽医专学报(2020年3期)2020-12-25
肉类研究(2020年9期)2020-12-14
世界科学技术-中医药现代化(2020年2期)2020-07-25
科技资讯(2020年3期)2020-04-07
中国现代神经疾病杂志(2020年1期)2020-01-08
中国中医急症(2019年10期)2019-05-21
肉类研究(2017年8期)2017-11-16
医学信息(2016年29期)2016-11-28
