
肢带型肌营养不良症2D型一家系临床表型及基因突变分析
2017-11-23 03:45欧俐羽孙毅明利婧王倞李欢曾缨梁颖茵张成
中国现代神经疾病杂志 2017年8期
欧俐羽 孙毅明 利婧 王倞 李欢 曾缨 梁颖茵 张成
·神经系统遗传性疾病·
肢带型肌营养不良症2D型一家系临床表型及基因突变分析
欧俐羽 孙毅明 利婧 王倞 李欢 曾缨 梁颖茵 张成
目的总结肢带型肌营养不良症2D型(LGMD2D型)临床表型和基因突变特点。方法报道一家系2例女性LGMD2D型患儿临床表现、肌电图、肌肉MRI、肌肉病理学和基因检测结果,并结合相关文献进行分析。结果先证者及其妹均于3岁发病,以进行性四肢近端无力为主要临床表现;血清肌酸激酶水平显著升高;肌电图呈肌源性损害;肌肉MRI显示部分肌肉萎缩、脂肪化或纤维水肿;其妹肌肉病理学显示局灶性骨骼肌坏死、再生,部分横纹肌消失,肌纤维大小不等。基因检测显示,先证者及其妹存在相同基因突变,即SGCA基因第3外显子移码突变c.262delT(p.Phe88SerfsX123)和第5外显子错义突变c.409G>A(p.Glu137Lys),其母为SGCA基因c.409G>A(p.Glu137Lys)突变携带者,其中,c.409G>A(p.Glu137Lys)为已知突变,c.262delT(p.Phe88SerfxX123)为新发突变。结论对于临床类似Duchenne型肌营养不良症的女性患者,排除DMD基因携带者后,还应行家系分析和肢带型肌营养不良症相关基因检测,以明确具体亚型。
肌营养不良,肢带型; 表型; 基因; 突变; 系谱
肢带型肌营养不良症2D型(LGMD2D型)是临床罕见的常染色体隐性遗传性肌营养不良症的一种亚型,通常于儿童期发病,临床表现为四肢近端无力、上楼梯和蹲起困难,血清肌酸激酶(CK)水平升高,肌电图呈肌源性损害,不易与Duchenne型肌营养不良症(DMD)区分,故亦称为类Duchenne型肌营养不良症肌病。目前,我国仅有1篇临床病理报告描述2例男性LGMD2D型患者[1],以及1篇文献报道台湾地区一家系共4例男性和1例女性LGMD2D型患者[2]。本研究报道一临床资料齐全家系共2例女性LGMD2D型患儿的家族史、临床表现、影像学、肌电图、肌肉病理学和基因突变特点,以期提高临床医师对LGMD2D型临床表型和基因突变的认识。
临床资料
一、临床特征
例1(先证者) 女性,8岁,主因四肢近端无力进行性加重5年,于2016年8月15日至我院神经科门诊就诊。患儿5年前无明显诱因出现四肢近端无力,跑步和上楼梯较同龄正常儿童缓慢,可平路行走,上肢可抬举,进行性加重,无感觉异常。外院肌电图检查显示,神经传导速度正常,呈肌源性损害。为求进一步诊断与治疗,遂至我院就诊。患儿足月剖宫产,出生时体重3.10 kg,无产伤和窒息史;智力发育正常,1岁时可独立行走,会说话;2岁时运动功能与同龄正常儿童无明显差异;3岁时跑步和上楼梯较同龄正常儿童缓慢;幼儿园(4~6岁)时跑步较同龄正常儿童缓慢,易疲劳、跌倒;7岁上小学,体育课上运动功能较同学落后,但无肌肉疼痛;8岁时行走缓慢,上楼梯和蹲起困难,需坐高凳子,可自行梳头,学习成绩中等。父母身体健康,无相似症状,否认近亲婚配。门诊体格检查:高级智能正常,脑神经检查未见异常;双上肢近端肌力4+级、远端5-级,双下肢近端肌力4级、远端5-级,肌张力均降低;双侧腓肠肌无肥大,但有韧性,无肌肉压痛,轻微腰椎前凸,无翼状肩胛、跟腱挛缩,可足尖行走,足跟行走欠佳,Gowers征阳性,可连跳3次,但跳不高;双上肢腱反射正常,双下肢膝反射和踝反射减弱,病理反射未引出。实验室检查:2016年8月15日血清肌酸激酶为 58 378 U/L(25~200 U/L),2017年3月13日血清肌酸激酶10 058 U/L、肌酸激酶同工酶(CK⁃MB)为150 U/L(2~24 U/L)、乳酸脱氢酶(LDH)518 U/L(114~240 U/L),α⁃羟丁酸脱氢酶(α⁃HBDH)275 U/L(80~220 U/L)、B型利尿钠肽(BNP)24.70 ng/L(0~84 ng/L)。心脏彩超显示心脏形态和结构无明显异常,心脏彩色多普勒超声(CDVS)无明显异常,左心室收缩和舒张功能正常。双侧大腿MRI检查(2017年3月20日)显示,大腿肌肉萎缩和脂肪化,以臀大肌、股二头肌、股四头肌和内收肌群为主,而缝匠肌、股薄肌、半腱肌相对保留(图1)。
例2(先证者之妹) 女性,3岁,因血清肌酸激酶升高1月余、四肢近端无力20 d余,于2013年8月26日首次至我院神经科门诊就诊。患儿于2013年7月曾因“咳嗽”住院,实验室检查血清肌酸激酶水平升高(2013年7月9日为8905 U/L和2013年7月11日为41 527 U/L),肌电图检查显示,双侧股四头肌肌源性损害。8月初家人发现其上楼梯困难,跑步较同龄正常儿童缓慢,可平路行走,上肢可抬举,无进行性加重,未见感觉异常。患儿足月顺产,出生时体重2.90 kg,无产伤和窒息史;智力发育正常,1岁时可独立行走;14个月时会说话;3岁时家人发现其上楼梯困难,跑步较同龄正常儿童缓慢。门诊体格检查:高级神经活动正常,脑神经检查未见异常;无明显肌萎缩,四肢近端肌力5-级、远端5级,肌张力均正常;双侧腓肠肌肥大,无肌肉压痛,平路行走正常,可足跟、足尖行走,可连跳;双上肢腱反射正常,双下肢膝反射减退、踝反射正常,病理反射未引出。实验室检查:2013年8月27日血清肌酸激酶12 215 U/L、肌酸激酶同工酶312 U/L、乳酸脱氢酶969 U/L、α⁃羟丁酸脱氢酶826 U/L,2016年8月15日血清肌酸激酶59 714 U/L、肌酸激酶同工酶371 U/L、乳酸脱氢酶1388 U/L。胸部X线和心电图未见明显异常。双下肢MRI检查(2015年9月22日)显示,广泛性脂肪浸润和纤维水肿,以臀大肌、比目鱼肌、腓肠肌为主(图2,3)。左侧腓肠肌肌肉组织活检可见局灶性骨骼肌坏死、再生,部分横纹肌消失,肌纤维大小不等。

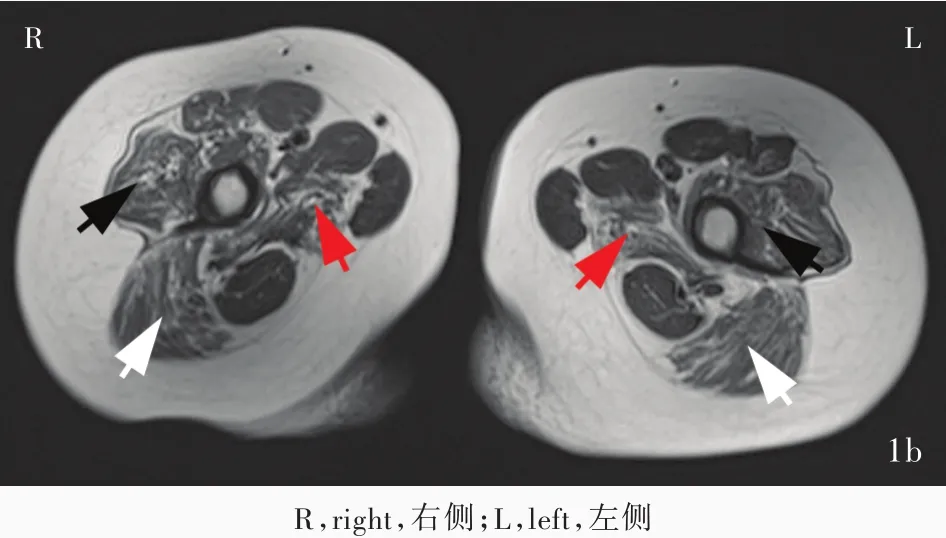
图1 双侧大腿MRI检查显示肌肉萎缩和脂肪化,以臀大肌(白箭头所示)、股四头肌(股外侧肌,黑箭头所示)和内收肌群(大收肌,红箭头所示)为主,缝匠肌、股薄肌和半腱肌形态和信号相对正常 1a 横断面T1WI 1b 横断面T2WI 1c 横断面抑脂T2WIFigure 1 Muscle MRI of the proband's thighs.Muscle of both thighs showed muscular atrophy and adipose infiltration mainly found in gluteus maximus (white arrows indicate),quadriceps(vastus lateralis,black arrows indicate)and adductors(adductor magnus,red arrows indicate).The shape and signal of sartorius,gracilis and semitendinosus were relatively normal. Axial T1WI(Panel 1a). Axial T2WI(Panel 1b). Axial fat saturation T2WI(Panel 1c).
二、基因检测
采用二代基因测序(NGS)技术对先证者(例1)外周静脉血样本进行肌肉病相关基因外显子捕获测序,结果显示,SGCA基因第3外显子存在移码突变 c.262delT(p.Phe88SerfsX123)和第 5外显子存在错义突变c.409G >A(p.Glu137Lys,图 4a)。进一步对先证者之母和妹c.262delT(p.Phe88SerfsX123)位点进行Sanger测序验证,结果显示,其母未携带该突变基因,其妹携带该突变基因(图4b);对c.409G>A(p.Glu137Lys)位点进行验证,结果显示,其母(图4c)和妹(图4b)均携带该突变基因。尽管先证者之父因故未行基因检测,但根据先证者及其母和妹的基因检测结果,推测移码突变c.262delT(p.Phe88SerfsX123)来自其父。采用Mutation Taster软件(http://www.mutationtaster.org/)预测上述两位点均为致病性突变,其中c.409G>A(p.Glu137Lys)是已报道的致病性突变,而c.262delT(p.Phe88SerfsX123)是移码突变,尚未见诸报道,为新发突变。经基因检测,先证者及其妹最终明确诊断为LGMD2D型。
三、家系分析
结合基因检测结果,得出该家系图,证实该家系为LGMD2D型常染色体隐性遗传性家系(图5)。
讨 论
肢带型肌营养不良症(LGMD)是以肩胛带肌、骨盆带肌、四肢近端肌肉不同程度进行性萎缩和无力为主要特点的一组肌肉病,根据遗传方式可以分为常染色体显性遗传性肢带型肌营养不良症(LGMD1型)和常染色体隐性肢带型肌营养不良症(LGMD2型);根据不同基因缺陷将LGMD1型和LGMD2型进一步分为多种亚型,目前已报道30余种亚型(http://www.musclegenetable.fr/),且具有高度遗传异质性。其中,LGMD2C、2D、2E和2F型分别与第 13、17、4和 5号染色体γ、α、β、δ⁃肌聚糖(sarcoglycan)编码基因突变有关,统称为肌聚糖病。γ、α、β、δ⁃肌聚糖组成一个跨膜的异四聚体,且与Sarcospan、抗肌萎缩蛋白(dystrophin)、肌营养不良蛋白聚糖(dystroglycan)、互生蛋白(syntrophin)和α⁃异联蛋白(α⁃dystrobrevin)形成抗肌萎缩蛋白⁃糖蛋白复合物(DGC)[3],该复合物在细胞外基底层、肌膜和细胞内骨架蛋白之间形成机械连接,在肌肉收缩和舒张过程中发挥稳定细胞膜的作用[4]。
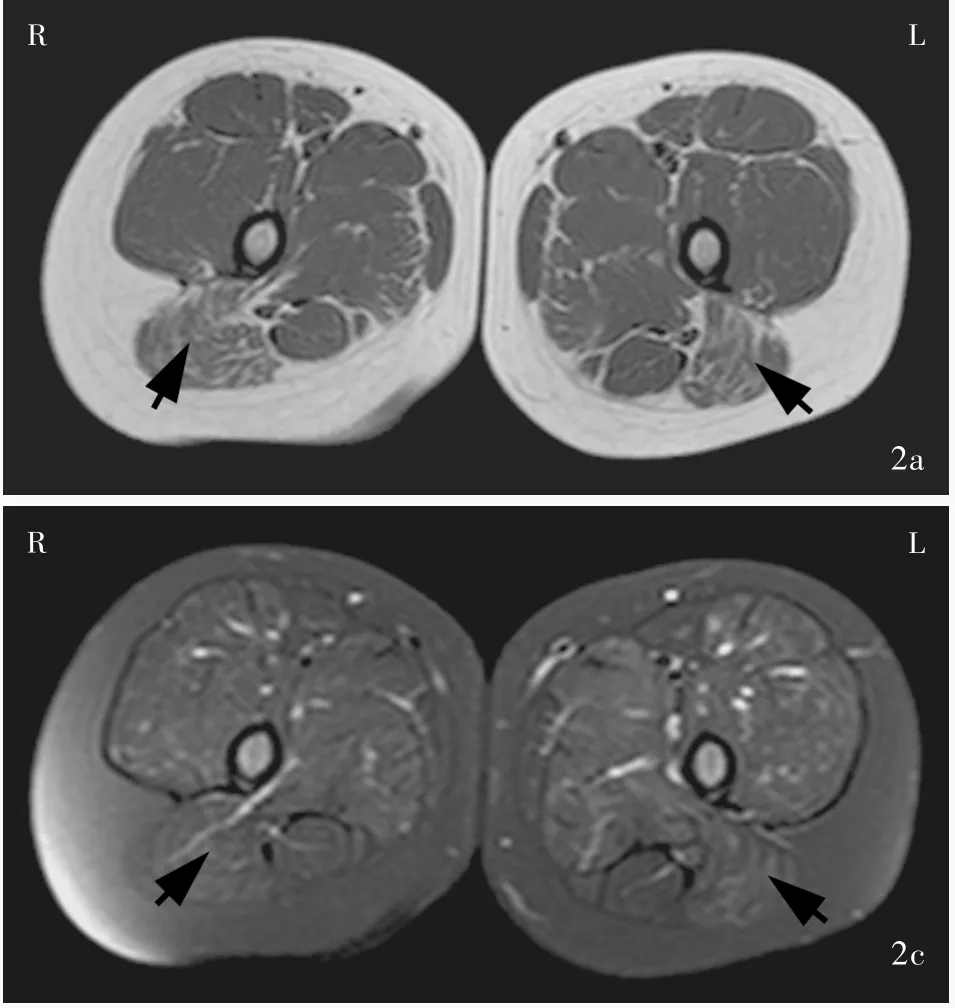

图2 双侧大腿MRI检查显示脂肪化和纤维水肿,以臀大肌为主(箭头所示) 2a 横断面T1WI 2b 横断面T2WI 2c 横断面抑脂T2WIFigure 2 Muscle MRI of thighs of the proband's younger sister. Fatnessand fiberedema were mainly found in gluteus maximus(arrows indicate).Axial T1WI(Panel 2a).Axial T2WI(Panel 2b).Axial fat saturation T2WI(Panel 2c).
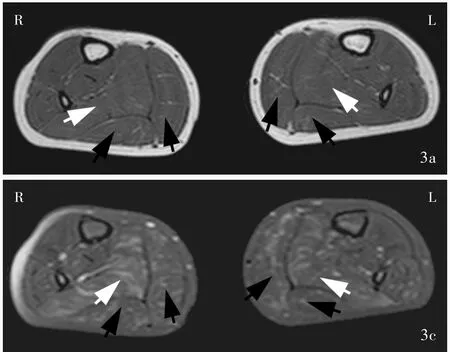

图3 双侧小腿MRI检查显示脂肪化和纤维水肿,以比目鱼肌(白箭头所示)和腓肠肌(黑箭头所示)为主 3a 横断面T1WI 3b横断面T2WI 3c 横断面抑脂T2WIFigure 3 Muscle MRI of calves of the proband's younger sister.Fatness and fiber edema were mainly found in soleus(white arrows indicate)and gastrocnemius(black arrows indicate). Axial T1WI(Panel 3a). Axial T2WI(Panel 3b). Axial fat saturation T2WI(Panel 3c).
LGMD2D型是肌聚糖病的最常见类型,由Roberds等[5]于 1994 年确定编码α⁃肌聚糖的 SGCA基因定位于17q12~q21.33,其错义突变导致α⁃肌聚糖缺陷引起的LGMD2D型。同年,McNally等[6]也将SGCA基因定位于17q21。SGCA基因包含10个外显子和2个启动子,α⁃肌聚糖cDNA由1410个核苷酸组成,其中开放阅读框包含1161个核苷酸,编码387 个氨基酸[7]。α⁃肌聚糖相对分子质量50×103,主要表达于骨骼肌,少量表达于心肌[8]。由于肌聚糖α、β、γ和δ 4个亚单位紧密连接,1个亚单位缺陷可以导致其他亚单位缺失,任意一种SGCA基因突变均可以引起部分或全部肌聚糖缺陷,甚至累及抗肌萎缩蛋白缺陷[9]。因此,免疫组织化学染色难以区分上述几种类型,明确诊断需借助基因诊断。


图4 SGCA基因检测所见 4a 先证者存在SGCA基因复合杂合突变c.262delT(p.Phe88SerfsX123,箭头所示,左图)和c.409G>A(p.Glu137Lys,箭头所示,右图) 4b 先证者之妹与先证者相同,存在SGCA基因复合杂合突变c.262delT(p.Phe88SerfsX123,箭头所示,左图)和c.409G>A(p.Glu137Lys,箭头所示,右图) 4c 先证者之母仅存在SGCA基因杂合突变c.409G>A(p.Glu137Lys,箭头所示)Figure 4 SGCA gene sequencing finding Compound heterozygous mutation c.262delT(p.Phe88SerfsX123,arrow indicates,left panel)and c.409G>A(p.Glu137Lys,arrow indicates,right panel)were seen in the proband(Panel 4a). The same SGCA gene sequencing results of the proband was seen in proband's younger sister(arrows indicate,Panel 4b).SGCA gene heterozygous mutation c.409G>A(p.Glu137Lys,arrow indicates)was seen in proband's mother(Panel 4c).
LGMD2D型与多种无义突变和错义突变有关,主要是SGCA基因第3外显子c.229C>T(p.Arg77Cys)突变,导致精氨酸突变为半胱氨酸[10⁃12]。本研究2例患儿均存在SGCA基因第3外显子移码突变 c.262delT(p.Phe88SerfsX123)和第 5外显子错义突变c.409G>A(p.Glu137Lys),其中c.409G>A(p.Glu137Lys)是已报道的致病性突变[10],而 c.262delT(p.Phe88SerfsX123)尚未见诸报道,从致病性突变分析原则看,可以认为系致病性突变。尽管先证者之父未行基因检测,但根据先证者及其母和妹的基因检测结果,可以推测移码突变c.262delT(p.Phe88SerfsX123)源自其父,这并不影响遗传信息学分析。
本研究家系患儿的可能发病机制为:SGCA基因第 3外显子存在 c.262delT(p.Phe88SerfsX123)碱基缺失,导致该基因编码的第88位密码子从苯丙氨酸突变为丝氨酸,并出现移码突变,提前出现终止密码子(第123位密码子),从而造成蛋白功能缺失;同时,SGCA基因第5外显子存在c.409G>A(p.Glu137Lys)碱基改变,导致该基因编码的第137位密码子由谷氨酸突变为赖氨酸,故可以确定2个突变位点位于2条染色体上,并经Mutation Taster软件预测均为致病性突变。2例患儿均存在SGCA基因移码突变c.262delT(p.Phe88SerfsX123)和错义突变c.409G > A(p.Glu137Lys),其 母 c.409G > A(p.Glu137Lys)为杂合突变,推测移码突变c.262delT(p.Phe88SerfsX123)可能源自其父,但患儿父母均无相似症状,推测其父亲可能为c.262delT(p.Phe88SerfsX123)杂合缺失,因此,该家系遗传方式呈常染色体隐性遗传。目前,关于SGCA基因移码突变c.262delT(p.Phe88SerfsX123)尚无相关报道,为新发突变,根据基因突变致病性判定标准[13],属致病性突变。已有文献报道,SGCA基因错义突变c.409G > A(p.Glu137Lys)具有致病性[10];此外,该密码子其他突变如c.410G>A(p.Glu137Gly)亦具有致病性[14]。
LGMD2D型主要临床特征为儿童期缓慢发病、四肢近端对称性肌萎缩和肌无力逐渐加重、呈“鸭步”步态,Gowers征阳性,不伴肌肉疼痛、肌强直,心肌受累少见[8,15],可累及呼吸肌,严重时可出现呼吸衰竭[8,16];血清肌酸激酶水平明显升高;肌电图和肌肉组织活检提示肌源性损害。本研究2例患儿均于3岁时出现上楼梯、蹲起、立位、跑步等运动功能较同龄正常儿童差,血清肌酸激酶水平高于正常参考值约50倍,肌电图和肌肉组织活检证实为肌源性损害,肌肉MRI显示广泛性脂肪化,基因检测为复合杂合突变,且在家系中出现共分离现象,复习相关文献,1种为已报道的致病性突变、1种为移码突变,故明确诊断为LGMD2D型。
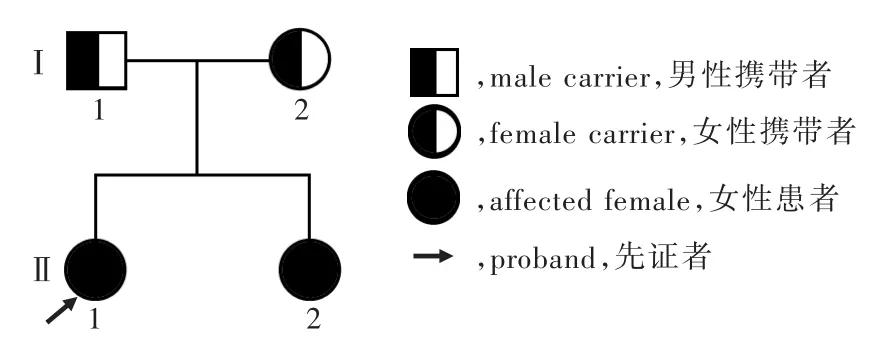
图5 LGMD2D型家系图Figure 5 The pedigree of LGMD2D.
LGMD2D型具有高度临床异质性,同一蛋白功能缺失,临床症状和病情严重程度不尽一致,甚至无明显临床症状。本研究2例患儿为同一种基因突变,先证者临床症状较其妹严重。1998年,Angelini等[17]报道2例LGMD2D型患者(一家系姊弟)均为同一种纯合突变,但临床表现截然不同:先证者为女性,40岁,于10~12岁发病,表现为轻度胸椎侧弯,28岁时出现下肢近端无力,30岁时出现上肢近端无力,Gowers征阳性,血清肌酸激酶1016 IU/L,超声心动图正常,肌电图提示肌源性损害,肌肉CT提示肌营养不良;其弟35岁,仅血清肌酸激酶水平升高(1037 IU/L),轻微脊柱侧弯,余未见异常。本研究2例患儿基因型相同,尽管先证者之妹较先证者症状轻微,但由于二者年龄相差较小(相差2岁),仍需进一步随访。
本研究先证者及其妹血清肌酸激酶水平明显升高,最高均超过50×103U/L,提示疾病处于活动期。研究显示,Duchenne型肌营养不良症和Becker型肌营养不良症(BMD)患者血清肌酸激酶水平较肢带型肌营养不良症患者高[18],前两者血清肌酸激酶可达正常参考值的25~200倍,后者为1~80倍,其中尤以LGMD2D型患者显著[19]。本研究2例患儿血清肌酸激酶水平升高,出现类似Duchenne型肌营养不良症表现,而先证者及其妹均无心脏受累,与心脏受累少见的报道相符[8,15]。先证者及其妹肌肉MRI均提示双下肢肌肉对称性受累,与临床表现相符。LGMD2D型肌肉MRI多表现为大腿前肌群较大腿后肌群更易受累[20]。先证者肌肉MRI显示大腿肌肉前后内侧肌群均受累,受累范围较广,而缝匠肌、股薄肌和半腱肌相对保留;其妹肌肉MRI显示以双下肢后肌群受累为主。随着肌肉MRI的普及,对可疑肌肉病的患者先行肌肉MRI检查,可以发现肌肉脂肪化和炎性水肿,结合肌肉受累顺序和病变程度,有助于判断肢带型肌营养不良症性质,并为肌肉组织活检部位的选择提供有效信息。
LGMD2D型临床表现类似Duchenne型肌营养不良症:先证者发病年龄较早,于3岁发病,四肢近端进行性肌无力,Gowers征阳性,血清肌酸激酶水平显著升高,高度怀疑女性Duchenne型肌营养不良症;其妹发病年龄也较早,于3岁发病,双侧小腿肥大,血清肌酸激酶水平显著升高,临床症状轻微,考虑DMD基因携带者。但二者本质上为LGMD2D型,与Duchenne型肌营养不良症的鉴别诊断要点为:性别不同;Duchenne型肌营养不良症为X连锁隐性遗传,多为男性;先证者无小腿肥大。因此,对于类似Duchenne型肌营养不良症的女性患者,除外DMD基因携带者,还应行家系分析和肢带型肌营养不良症相关基因检测,以明确具体分型。
由于肢带型肌营养不良症具有高度遗传异质性,故在临床上难以区分具体亚型,明确诊断主要依靠基因检测。然而某些临床特征可以缩小鉴别诊断范围,有助于基因检测包的选择。例如,女性肢带型肌营养不良症应注意与女性Duchenne型肌营养不良症相鉴别,二者均表现为儿童期近端肌萎缩和肌无力、血清肌酸激酶水平明显升高,但前者呈常染色体隐性或显性遗传,症状较后者严重,肌肉组织活检抗肌萎缩蛋白阴性;后者呈X连锁隐性遗传,肌肉组织活检抗肌萎缩蛋白部分阳性。此外,肢带型肌营养不良症还应注意与炎性肌病、代谢性肌病相鉴别:均表现有近端肌无力,血清肌酸激酶水平升高,肢带型肌营养不良症起病隐匿、进展缓慢,不伴肌肉疼痛;炎性肌病无家族史,起病急骤,进展迅速,可伴肌肉疼痛,肌电图多呈活动性肌源性损害,可见较多正尖纤颤波,糖皮质激素治疗有效,可资鉴别;代谢性肌病方面主要是脂质沉积性肌病,病程呈波动性,肌肉组织活检可见肌纤维内堆积大量脂肪滴,而肢带型肌营养不良症病程无波动,肌肉组织活检提示肌纤维破坏,无脂肪滴堆积,可资鉴别。
[1]Song XQ,Yao MM,Wu HR,Liu LL,Wang X,Li W,Chen XX.Clinicaland pathologicalfeatures ofα ⁃Sarcoglycanopathy(report of 2 cases).Lin Chuang Shen Jing Bing Xue Za Zhi,2015,28:131⁃133[.宋学琴,要萌萌,吴红然,刘璘琳,王雪,李薇,陈秀晓.α⁃Sarcoglycanopathy的临床和病理学特征:附2例报告.临床神经病学杂志,2015,28:131⁃133.]
[2]Liang WC,Chou PC,Hung CC,Su YN,Kan TM,Chen WZ,Hayashi YK,Nishino I,Jong YJ.Probable high prevalence of limb⁃girdle muscular dystrophy type 2D in Taiwan.J Neurol Sci,2016,362:304⁃308.
[3]Rando TA.The dystrophin⁃glycoprotein complex,cellular signaling,and the regulation of cell survival in the muscular dystrophies.Muscle Nerve,2001,24:1575⁃1594.
[4]Ervasti JM,Campbell KP.A role for the dystrophin⁃glycoprotein complex as a transmembrane linker between laminin and actin.J Cell Biol,1993,122:809⁃823.
[5]Roberds SL,Leturcq F,Allamand V,Piccolo F,Jeanpierre M,AndersonRD,Lim LE,LeeJC,ToméFM,RomeroNB,Fardeau M,Beckmann JS,Kaplan JC,Campbell KP.Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy.Cell,1994,78:625⁃633.
[6]McNally EM,Yoshida M,Mizuno Y,Ozawa E,Kunkel LM.Human adhalin is alternatively spliced and the gene is located on chromosome 17q21.Proc Natl Acad Sci USA,1994,91:9690⁃9694.
[7]Ozawa E,Mizuno Y,Hagiwara Y,Sasaoka T,Yoshida M.Molecular and cell biology of the sarcoglycan complex.Muscle Nerve,2005,32:563⁃576.
[8]Politano L,Nigro V,Passamano L,Petretta V,ComiLI,Papparella S,Nigro G,Rambaldi PF,Raia P,Pini A,Mora M,Giugliano MA,Esposito MG,Nigro G.Evaluation of cardiac and respiratory involvement in sarcoglycanopathies.Neuromuscul Disord,2001,11:178⁃185.
[9]VainzofM,Passos⁃BuenoMR,CanovasM,MoreiraES,Pavanello RC,Marie SK,Anderson LV,Bonnemann CG,McNally EM,Nigro V,Kunkel LM,Zatz M.The sarcoglycan complex in the six autosomal recessive limb⁃girdle muscular dystrophies.Hum Mol Genet,1996,5:1963⁃1969.
[10]Carrié A,Piccolo F,Leturcq F,de Toma C,Azibi K,Beldjord C,Vallat JM,Merlini L,Voit T,Sewry C,Urtizberea JA,Romero N,Tomé FM,Fardeau M,Sunada Y,Campbell KP,Kaplan JC,Jeanpierre M.Mutational diversity and hot spots in the alpha⁃sarcoglycan gene in autosomal recessive muscular dystrophy(LGMD2D).J Med Genet,1997,34:470⁃475.
[11]Hackman P,Juvonen V,Sarparanta J,Penttinen M,Aärimaa T,Uusitalo M,Auranen M,Pihko H,Alén R,Junes M,Lönnqvist T,Kalimo H,Udd B.Enrichmentofthe R77C alpha⁃sarcoglycan gene mutation in Finnish LGMD2D patients.Muscle Nerve,2005,31:199⁃204.
[12]Trabelsi M,Kavian N,Daoud F,Commere V,Deburgrave N,Beugnet C,Llense S,Barbot JC,Vasson A,Kaplan JC,Leturcq F, Chelly J. Revised spectrum of mutations in sarcoglycanopathies.Eur J Hum Genet,2008,16:793⁃803.
[13]Richards S,Aziz N,Bale S,Bick D,Das S,Gastier⁃Foster J,Grody WW,Hegde M,Lyon E,Spector E,Voelkerding K,Rehm HL;ACMG Laboratory Quality Assurance Committee.Standards and guidelines for the interpretation of sequence variants:a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.Genet Med,2015,17:405⁃424.
[14]Kawai H,Akaike M,Endo T,Adachi K,Inui T,Mitsui T,Kashiwagi S,Fujiwara T,Okuno S,Shin S.Adhalin gene mutations in patients with autosomal recessive childhood onset musculardystrophy with adhalin deficiency.JClin Invest,1995,96:1202⁃1207.
[15]Fayssoil A,Nardi O,Annane D,Orlikowski D.Left ventricular function in alpha⁃sarcoglycanopathy and gamma⁃sarcoglycanopathy.Acta Neurol Belg,2014,114:257⁃259.
[16]WalterMC,DekomienG,Schlotter⁃WeigelB,ReilichP,Pongratz D,Müller⁃FelberW,Epplen JT,HuebnerA,Lochmüller H. Respiratory insufficiency as a presenting symptom of LGMD2D in adulthood.Acta Myol,2004,23:1⁃5.
[17]Angelini C,Fanin M,Menegazzo E,Freda MP,Duggan DJ,Hoffman EP.Homozygous alpha⁃sarcoglycan mutation in two siblings:one asymptomatic and one steroid⁃responsive mild limb⁃girdle muscular dystrophy patient.Muscle Nerve,1998,21:769⁃775.
[18]Zhang Y,Huang JJ,Wang ZQ,Wang N,Wu ZY.Value of muscle enzyme measurement in evaluating different neuromuscular diseases.Clin Chim Acta,2012,413(3/4):520⁃524.
[19]Passos⁃Bueno MR,Vainzof M,Moreira ES,Zatz M.Seven autosomal recessive limb⁃girdle muscular dystrophies in the Brazilian population:from LGMD2A to LGMD2G.Am J Med Genet,1999,82:392⁃398.
[20]Wattjes MP,Kley RA,Fischer D.Neuromuscular imaging in inherited muscle diseases.Eur Radiol,2010,20:2447⁃2460.
Limb⁃girdle muscular dystrophy type 2D:clinical and genetic analysis of a family
OU Li⁃yu1,SUN Yi⁃ming2,LI Jing1,WANG Liang1,LI Huan1,ZENG Ying1,LIANG Ying⁃yin1,ZHANG Cheng11Department of Neurology,2Department of Health Care Clinic,the First Affiliated Hospital,Sun Yat⁃sen University,Guangzhou 510080,Guangdong,China
OU Li⁃yu and SUN Yi⁃ming contributed equally to this study
Corresponding author:ZHANG Cheng(Email:zhangch6@mail.sysu.edu.cn)
Objective To study the characteristics and diagnosis of limb⁃girdle muscular dystrophy type 2D(LGMD2D).Methods The clinical characteristics,EMG,muscle MRI and muscle pathological studies of 2 female patients in a family with LGMD2D were analyzed.Genetic analysis was used in the diagnosis of this disease.The cases were reported along with related literatures review.Results The onset of the proband and her younger sister occurred at 3 years old with progressive proximal muscle weakness of four limbs as the main clinical manifestation.The serum creatine kinase(CK)was significantly high(> 50 × 103U/L).EMG showed myogenic damage.Muscle MRI indicated partial muscle atrophy,fatness or fiber edema.Muscle pathological examination of the proband's younger sister revealed skeletal muscle necrosis and focal regeneration,partial striated muscle disappearance,and the muscle fibers in different sizes.Sequencing of all 10 coding exons of the SGCA gene in 2 patients revealed the same mutation:a c.262delT(p.Phe88SerfsX123)frameshift mutation in exon 3 and a c.409G>A(p.Glu137Lys)missense mutation in exon 5.Their mother was a carrier of SGCA gene c.409G>A(p.Glu137Lys)mutation.c.409G>A (p.Glu137Lys)is a mutation already found,and c.262delT(p.Phe88SerfsX123)is a novel mutation.The proband's father did not take the genetic test for some reason.Conclusions In case of a female with Duchenne muscular dystrophy(DMD)⁃like symptom,if she has been excluded from the DMD gene carrier,pedigree analysis and genetic analysis involving limb⁃girdle muscular dystrophy(LGMD)should be conducted to facilitate the diagnosis of the LGMD and its subtypes.
Muscular dystrophies,limb⁃girdle; Phenotype; Genes; Mutation; Pedigree
10.3969/j.issn.1672⁃6731.2017.08.010
欧俐羽、孙毅明并列为本文第一作者
国家自然科学基金资助项目(项目编号:81471280);广东省广州市2015年产学研专项项目(项目编号:1561000153);国家自然科学基金青年科学基金资助项目(项目编号:81601087);广东省科学技术厅2014年度公益研究与能力建设专项资金资助项目(项目编号:2014A020212130);国家自然科学基金资助项目(项目编号:81271401);国家自然科学基金-广东省联合基金重点资助项目(项目编号:U1032004)
510080广州,中山大学附属第一医院神经科[欧俐羽(现在广西中医药大学附属瑞康医院神经内科,邮政编码:530011)、利婧、王倞、李欢、曾缨、梁颖茵、张成],保健科(孙毅明)
张成(Email:zhangch6@mail.sysu.edu.cn)
This study was supported by the National Natural Science Foundation of China(No.81471280,81271401),2015 Production,Study and Research Special Project of Guangzhou,Guangdong Province,China(No.1561000153),the National Natural Science Foundation of China for Young Scientists(No.81601087),Non⁃Profit Study and Capability Building Special Fund Support Project of Guangdong Provincial Department of Science and Technology,China in the Year 2014(No.2014A020212130),and Joint Fund of National Natural Science Foundation of China and Natural Science Foundation of Guangdong Province,China(No.U1032004).
2017⁃06⁃29)
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
临床输血与检验(2022年3期)2022-06-22
山东医药(2021年24期)2021-09-01
冰雪运动(2021年1期)2021-07-28
中国生殖健康(2020年4期)2021-01-18
安徽医专学报(2020年3期)2020-12-25
诊断学(理论与实践)(2020年1期)2020-04-28
郑州大学学报(医学版)(2019年3期)2019-06-03
中国生殖健康(2018年4期)2018-11-06
医学信息(2016年29期)2016-11-28
