
硼氮掺杂C64-石墨炔材料几何及电子结构第一性原理
2020-12-10 02:09辛子华
上海大学学报(自然科学版) 2020年5期
李 晖, 辛子华
(上海大学理学院, 上海200444)
碳的各种同素异形体一直是科研工作者研究的焦点, 其中石墨烯自2004 年成功合成[1]以来, 因其优异性能成为新的研究热点. 越来越多的科学家开始投入石墨烯的研究中, 如对石墨烯的电子结构[2]、光学透明度[3]等性质进行研究. 但由于石墨烯是零带隙材料, 在半导体器件中的应用受到局限, 比如无法制成场效应管等. 因此, 研究者开始通过掺杂[4]或分割成带结构[5]等方法, 对石墨烯带隙进行调制. 已有研究表明, 硼、氮、氧和氟原子掺杂石墨烯均能有效改变石墨烯的带隙, 并且氟原子的掺杂使结构带有磁性[4]. 目前, 硼和氮掺杂的石墨烯结构已被成功合成[6-8]. 寻找更多碳的同素异形体结构, 获取新的特殊性能, 成为重要研究方向.
目前较引人注目的是石墨炔结构, 最早由Baughman等[9]于1987 年在理论上提出. 直到2009 年, 石墨炔才由Li等[10]利用六炔基苯在铜的催化作用下合成出来, 即石墨二炔. 石墨二炔的合成让人们看到了应用炔材料的可能性, 其后开展了很多对石墨炔及其衍生物的理论研究工作. Zhang等[11]发现 α-, β-, γ-以及 6,6,12-石墨炔的力学性能具有明显的各向异性.Long等[12]发现石墨二炔具有较高的电子迁移率, 其带隙宽度为0.45 eV. γ-石墨炔及其二炔结构具有良好的热稳定性能[13]. Zhang等[14]和Liu等[15]的研究表明, 锗原子的掺杂能有效调节α-石墨炔、γ-石墨炔及其相关结构的带隙. 硅掺杂石墨二炔形成的碳硅二炔结构在很大温度范围内能保持稳定性[16], 并且当达到一定温度后, 六元环附近开始出现碳硅四元环, 且将温度降回0 K 时, 碳硅四元环不会消失. 此外, 硼[17-19]、氮[17-18]、氧[18]和铝[19]等原子的掺杂都能有效调节石墨炔的带隙. 原胞结构为平行四边形结构的石墨炔在2015 年出现[20], 是带隙为0 eV、具有狄拉克锥的单原子层平面材料.
Song等[21]对同时含有碳四元环和六元环的碳的同素异形体——Graphenylene 结构进行了研究. Graphenylene 结构中的六元环属于环几三烯结构单元(cyclohexatriene), 该单元中存在两种不同的碳碳键, 键长分别为1.366×10-10和1.467×10-10m. 计算表明, 这种含有环几三烯单元的结构较γ-石墨炔和卡拜更为稳定. Lu等[22]研究了4 种碳同素异形体C65, C63,C31和C41结构, 它们分别由碳六元环和五元环、碳六元环和三元环、碳三元环和一个碳原子、碳四元环和一个碳原子构成, 这4 种结构均具有金属性. 已有研究表明, 构成石墨炔的基本单元具有多样性, 这种结构单元的多样性及结构单元间的不同连接方式使得石墨炔结构具有多样性. 在前期研究中, 本团队获得了一种新型的石墨炔结构, 即C64-石墨炔, 它含有碳的六元环和四元环以及碳碳三键(炔键), 其中的碳六元环含有两种非离域的碳碳键, 也属于环几三烯单元结构. C64-石墨炔的晶格常数为9.291×10-10m, 是具有带隙宽度为0.35 eV 的直接带隙半导体. 本工作计算了3 种C64-石墨炔的掺杂结构, 这3 种结构都是单原子层平面材料.
1 计算方法
本工作采用基于密度泛函理论(density functional theory, DFT)的第一性原理计算方法,借助Vienna Ab-initio Simulation Package (VASP)软件包对3 种单原子层平面结构进行研究. 在计算过程中, 采用缀加平面波(projected augmented wave, PAW)方法处理电子-离子相互作用, 利用广义梯度近似(generalized gradient approximation, GGA)下的Perdew-Burke-Ernzerhof(PBE)方法对交换关联泛函进行处理. 所有结构的计算均采用550 eV 的平面波截断能, 能量和力的收敛精度分别设为1×104和1×107eV·m, K 点网格取为11×11×1, 为避免层间相互作用, 真空层选取为2×10-9m. 对优化后的结构还进行了Γ 点的声子计算以及电子结构的计算.
2 结果与讨论
2.1 结构表征
为探讨硼、氮原子取代C64-石墨炔中的碳原子后所得结构的特点, 分别对不同掺杂位置、不同掺杂数量的十几种几何结构进行弛豫计算, 发现当用单个硼原子取代C64-石墨炔碳四元环中的一个碳原子或碳六元环中的一个碳原子时, 得到两种单原子层平面结构, 分别记为四元环位B 掺杂C64-石墨炔结构和六元环位B 掺杂C64-石墨炔结构, 简称B4 环掺杂结构(B4ringC-C64)和B6 环掺杂结构(B6ringC-C64); 当用硼和氮原子交替取代四元环和碳链上的碳原子时, 获得一种单原子层平面结构, 记为链位B, N 掺杂C64-石墨炔, 简称BN 链掺杂结构((BN)chain-C64). 这几种结构在图1 中给出, 图中红色平行四边形圈起来的部分为优化所选取的原胞; 灰色、粉色和蓝色球形分别为碳原子、硼原子和氮原子. 图1(a)是前期研究中得到的C64-石墨炔, (b)~(d)分别为B4 环掺杂结构、B6 环掺杂结构和BN 链掺杂结构. 这4 种结构优化后的晶格常数和能量在表1 中给出. 与C64-石墨炔相比, 掺杂后3 种结构的晶格常数均变大. 另外, 由于B4 环掺杂结构的总能低于B6 环掺杂结构, 说明硼原子掺在四元环比掺在六元环上结构更为稳定.

图1 晶格结构Fig.1 Structures of lattice

表1 C64-石墨炔、B4 环掺杂结构、B6 环掺杂结构和BN 链掺杂结构的晶格常数和总能Table 1 Lattice constant and total energy of C64-graphyne, B4ringC-C64, B6ringC-C64 and (BN)chain-C64 structure
C64-石墨炔是一种同时含有碳六元环和碳四元环的独特结构, 而其中的碳六元环存在两种不同碳碳键长的环几三烯单元, 这与已知石墨炔中以苯环形式存在的碳六元环不同. 在这种碳六元环中, 两种不同键长的碳碳键交替存在, 键长分别为1.378×10-10和1.460×10-10m.同时, 碳链上有一个由sp 杂化碳原子形成的炔键, 键长为1.253×10-10m. 研究结果表明, 利用硼原子掺杂后, 所得的B4 环掺杂结构虽略有畸变, 但其六元环仍然表现为长短交替, 称为类环己三烯结构; 而B6 环掺杂结构由于硼碳键较长, 使其六元环结构中的键长不再是长短交替.两种结构碳链上仍然存在炔键, 键长分别为1.254×10-10和1.249×10-10m; 利用硼原子和氮原子交替取代四元环和碳链上的碳原子, 得到的BN 链掺杂结构中仍然存在环己三烯单元, 其链上出现了硼、氮原子形成的炔键, 键长为1.281×10-10m.
为考证B4 环掺杂结构、B6 环掺杂结构和BN 链掺杂结构的稳定性, 计算了这3 种结构在Γ 点的声子频率. 3 种结构原胞均有18 个原子, 声子计算分别得到54 支频率. 结果表明, 所有光学支谱线在Γ 点均为正值; 3 支声学支谱线在Γ 点虽然有负值, 但其绝对值均小于0.1 THz,属于计算误差. Γ 点声子计算结果进一步表明这3 种掺杂方式仍能保证材料结构的稳定性.
2.2 电子结构
本工作对上述得到的3 种单原子层材料的电子结构进行了计算. 能带结构如图2 所示,其中图(a)是为了进行比较而给出的C64-石墨炔能带结构, (b)~(d)分别对应B4 环掺杂结构、B6 环掺杂结构和BN 链掺杂结构的能带图. 为了比较4 种结构的电子结构特性, 还在表2 中给出了C64-石墨炔、B4 环掺杂结构、B6 环掺杂结构和BN 链掺杂结构的带隙宽度以及带隙出现的位置. C64-石墨炔是一种带隙值为0.35 eV 的直接带隙半导体, 其价带顶和导带底均出现在M 点处. 掺杂后的B4 环掺杂结构和B6 环掺杂结构的能带图中费米能级附近均有一条较宽的能带穿过费米能级, 结合它们的态密度(density of states, DOS)图(见图3 和4),可知这两种结构均表现出金属性质. 图3 和4 中Total DOS 代表总态密度, LDOS 代表局域态密度(local DOS), PDOS 代表分波态密度(partial DOS). 此外, BN 链掺杂结构的带隙为2.56 eV, 为间接带隙半导体, 其价带顶出现在Γ 点, 导带底出现在M 点, 费米能级附近能带宽度较窄.
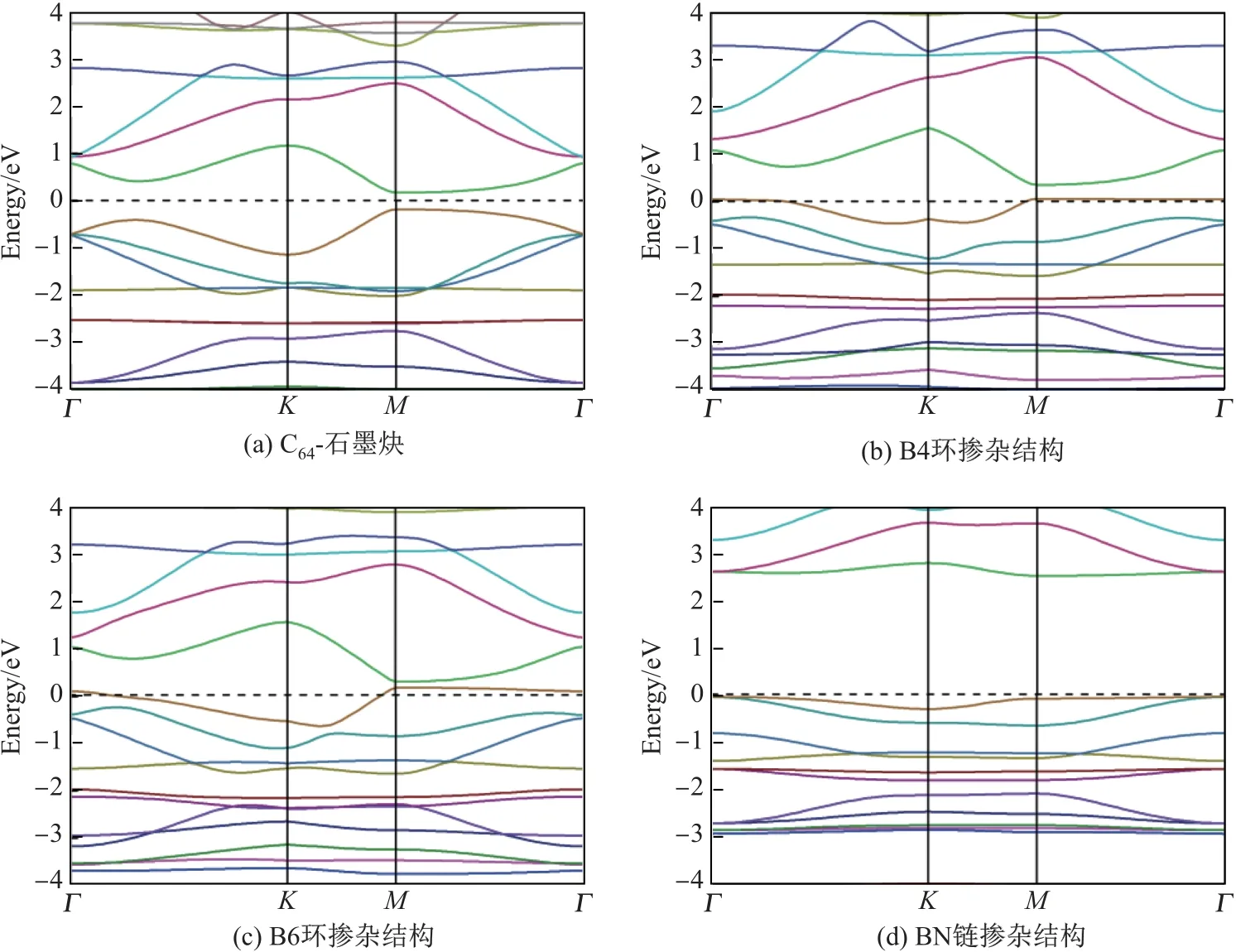
图2 能带结构Fig.2 Band structures

表2 C64-石墨炔、B4 环掺杂结构、B6 环掺杂结构和BN 链掺杂结构的带隙宽度与位置Table 2 Band gaps and positions of C64-graphyne, B4ringC-C64, B6ringC-C64 and(BN)chain-C64 structure
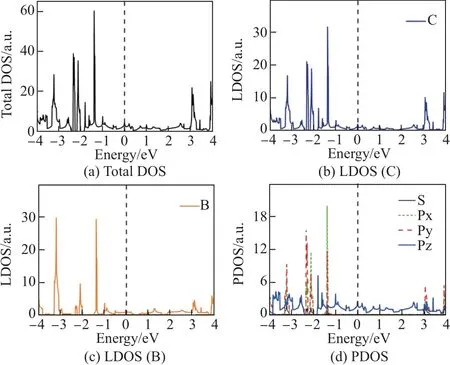
图3 B4 环掺杂结构态密度图Fig.3 Densities of states of B4ringC-C64 structure
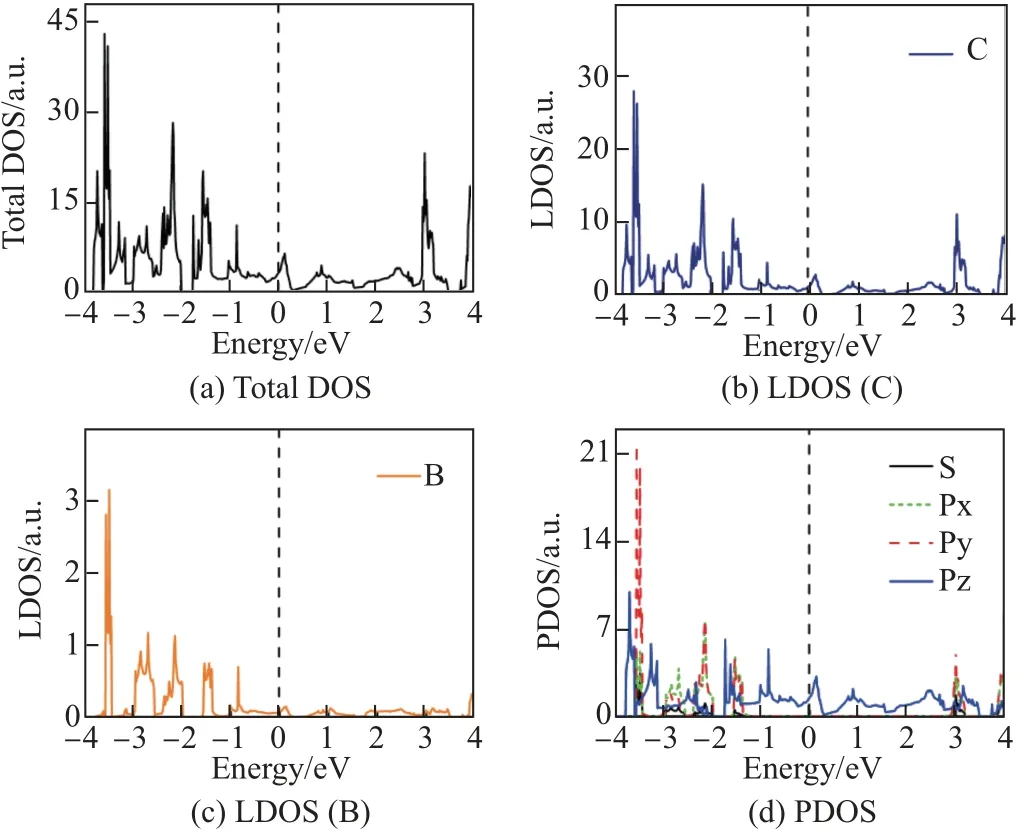
图4 B6 环掺杂结构态密度图Fig.4 Densities of states of B6ringC-C64 structure
为研究3 种掺杂结构中各原子以及各轨道电子对体系电子结构的贡献, 对3 种结构的总态密度、局域态密度和分波态密度进行了计算(见图3~5). 为便于比较, C64-石墨炔的总态密度和分波态密度在图6 中给出. 由图3 和4 中局域态密度可知, 在能量为0 处, 硼原子态密度基本为0, B4 环掺杂结构和B6 环掺杂结构在费米面附近电子结构几乎完全由碳原子贡献. 而硼原子的贡献在于改变了碳原子的态密度分布,使得碳原子态密度由图6 所示的在费米面处存在带隙的结构, 改变为无带隙的结构, 这也是使C64-石墨炔在掺杂一个硼原子时表现为金属性的原因. 由图3 和4 中的PDOS 曲线可以看出, B4 环掺杂结构和B6 环掺杂结构在费米能级附近处于S, Px 和Py 轨道的电子对费米能级附近的态密度也几乎没有贡献, 这与图6 所示的费米能级附近的态密度主要由碳原子中处于Pz 轨道的电子贡献的结果是相同的. 由图5(b)和(c)可知, BN 链掺杂结构中的B, N 原子以及C 原子对费米面附近的电子态密度均有贡献.
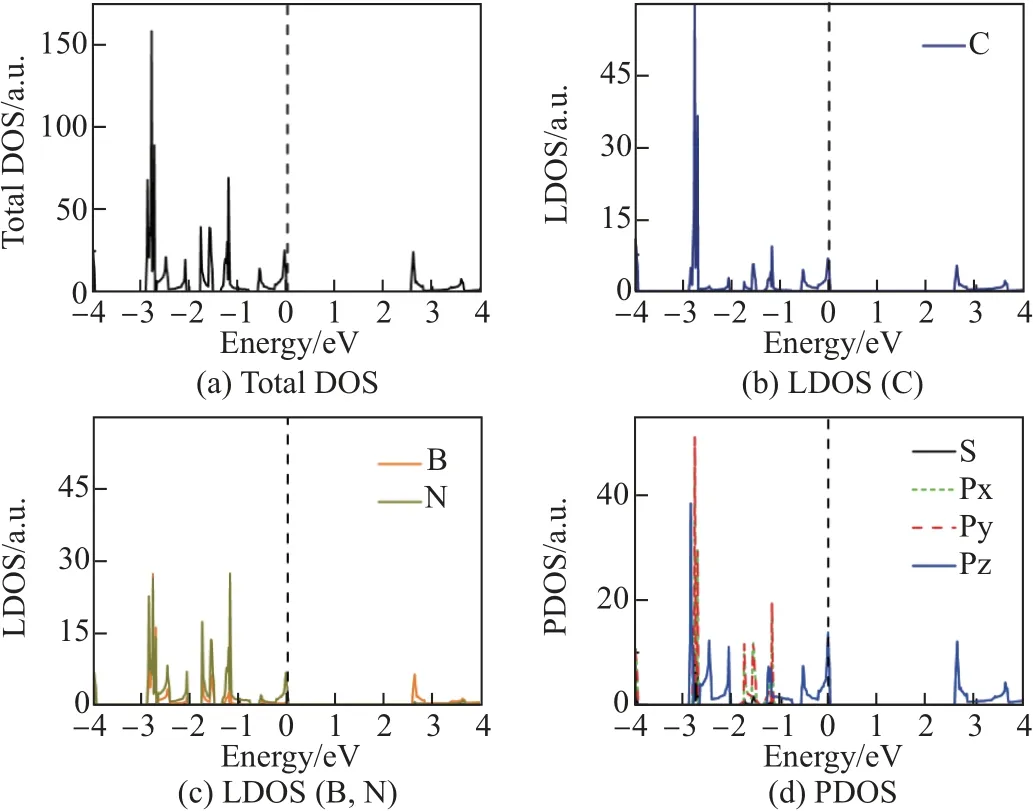
图5 BN 链掺杂结构态密度图Fig.5 Densities of states of (BN)chain-C64 structure

图6 C64-石墨炔态密度图Fig.6 Densities of states of C64-graphyne structure
比较图5 和6 中的碳原子态密度可知, 由于硼, 氮原子的引入使得费米面附近碳原子带隙展宽, 这也导致硼, 氮同时掺杂的BN 链掺杂结构带隙变宽. 同时, BN 链掺杂结构价带顶和导带底附近电子的总态密度存在尖锐的峰, 说明处于价带顶和导带底附近的电子态密度比C64-石墨炔大. 由图5 中的局域态密度还可知, 硼原子和碳原子对价带附近的态密度贡献较大, 而氮原子和碳原子对导带附近的态密度贡献较大; 结合PDOS 曲线可知, 导带底附近电子态密度主要由氮原子和碳原子中处于Pz 轨道的电子贡献, 价带顶附近电子态密度主要由硼原子和碳原子中处于Pz 轨道的电子贡献.
3 结束语
本工作主要研究了3 种稳定的单原子层平面材料, 即B4ringC-C64, B6ringC-C64和(BN)chain-C64, 包括它们的几何结构、稳定性和电子结构. 单个硼原子的掺杂得到两种单原子层平面结构: B4 环掺杂结构和B6 环掺杂结构. B4 环掺杂结构的六元环为类环几三烯单元, 环上的6 个键的键长长短交替, 但不再只含有完全相同的两种键长. 两种结构的碳链上仍然存在一个由sp 杂化的碳原子构成的炔键, 键长分别为1.254×10-10和1.249×10-10m. 硼原子的掺杂使得这两种结构均具有金属性质, 且硼原子掺杂到六元环上较掺杂到四元环上更为稳定. 硼、氮原子交替取代C64-石墨炔中四元环和碳链上的碳原子, 仍然能获得一种单原子层平面结构, 这种BN 链掺杂结构的六元环中仍以环己三烯单元的形式存在, 有长短交替的两种键长, 分别为1.373×10-10和1.446 ×10-10m. 电子结构的计算结果表明, BN 链掺杂结构是具有带隙为2.56 eV 的半导体. 3 种单原子层平面结构的不同性能决定了它们在不同领域潜在的应用价值, 比如3 种结构中含有多种大小不同的孔洞, 最大的环为十二元环, 可以考虑作为气体的分离材料.
猜你喜欢
原子与分子物理学报(2022年3期)2022-03-05
辽宁科技大学学报(2021年2期)2021-07-22
中小学班主任(2019年12期)2019-09-10
青岛大学学报(工程技术版)(2019年2期)2019-09-10
科技创新与应用(2018年21期)2018-09-14
振动工程学报(2017年4期)2018-05-31
电子制作(2018年1期)2018-04-04
新课程·中旬(2016年12期)2017-05-08
振动工程学报(2017年1期)2017-04-21
中学生数理化·高二版(2017年2期)2017-04-19
