
5/7型猪细小病毒双重PCR检测方法的建立
2021-09-10 05:45王茂鹏王伟伟李乐天鲁会军金宁一米志强
中国兽医学报 2021年8期
王茂鹏,王伟伟,李 笨,李乐天,鲁会军,金宁一,韩 硕,米志强,李 昌*
(1.温州大学 病毒学研究所,浙江 温州 325035;2.军事科学院 军事医学研究院 军事兽医研究所 中国医学科学院人兽共患病毒病防控关键技术研究创新单元,吉林 长春 130122;3.辽宁省农业发展服务中心,辽宁 沈阳 110031;4.军事科学院 军事医学研究院 微生物流行病研究所,北京100071)
猪细小病毒(porcine parvoviruses,PPV)为细小病毒科细小病毒属成员,可引起母猪木乃伊胎、不孕不育及死胎等繁殖障碍性疾病,在世界各地的猪群中均有发现,是造成全球养猪业经济损失的重要传染病之一[1-5]。近年来,已发现7个基因型PPV(1~7),通常用于区分猪细小病毒亚科成员,而这7个基因型PPV根据NS1蛋白序列同源性在细小病毒系统发育树上又被划分为Protoparvovirus、Tetraparvovirus、Chapparvovirus和Copiparvovirus4个不同的属[5-7]。
PPV5和PPV7分别属于Copiparvovirus属和Chapparvovirus属,于2013和2016年才被相继发现。XIAO等[4]对美国不同年龄段猪群感染PPV5进行的流行病学调查结果显示,其阳性率达6.6%,略高于PPV4发病率(4.1%)。除美国外,在中国、波兰和墨西哥等国家也陆续检出PPV5,PPV5常与PPV4及猪圆环病毒发生共感染,在肺组织内有较高的检出率,可造成严重的仔猪断奶后多系统衰弱综合征(post-weaning multi-systemic wasting syndrome,PMWS)[3,8-11]。PPV7亚型细小病毒已经在我国东北三省、广西省、安徽省等多地检出。现有文献表明,PPV5和PPV7共感染加重猪只病情[12-14],但由于这2个亚型发现时间较短,对共感染病例的检测尚缺乏有效方法,本研究建立一种针对PPV5/PPV7的双重PCR检测方法,并对其进行优化,以期为深入了解PPV共感染对宿主状态的影响储备关键技术。
1 材料与方法
1.1 主要试剂和仪器E.coliDH5α感受态细胞、pBM16A载体购自博迈德生物有限公司;LAMP酶、2×Taq DNA Master Mix购自南京诺维赞生物科技有限公司;KOD酶购自日本TOYOBO生物公司;NanoDrop ND-2000C微量核酸检测仪购自美国Thermo Fisher生物公司;ABI Veriti 96孔梯度PCR仪购自ABI公司。
1.2 引物设计为了同时满足PPV5/7的特异性检测和基因组测序分型,以NCBI数据库中PPV5/7序列NS1基因保守区域为模板(https://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi GROUP_TARGET=on)设计2对引物(表1),由生工生物工程(上海)股份有限公司合成。

表1 双重PCR引物序列
1.3 扩增目的片段按照病毒基因组DNA/RNA提取试剂盒使用说明书提取病毒核酸,-80℃保存备用。以提取的病毒DNA为模板,用表1中相应的引物分别进行PCR扩增,回收扩增产物连接至pBM16A载体,转化至E.coliDH5α感受态细胞,接种LB氨苄抗性液体培养基培养12 h,提取质粒,经通用引物和特异性引物鉴定条带大小正确后,送至生工生物工程(上海)股份有限公司测序鉴定。
1.4 核酸模板定量、稀释与标准品制备以测序正确的重组质粒作为标准品,用Nano-Drop2000C超微量分光光度计测定浓度后,根据公式[拷贝数=质粒浓度×10-9×6.02×1023/(660×质粒总长度)]将其换算为拷贝数,10倍倍比稀释成终浓度为1.0×1013~1.0×101拷贝/μL,作为检测标准品。
1.5 双重PCR敏感性试验分别取稀释至1.0×101~1.0×108拷贝/μL的重组质粒标准品并等体积混合,作为双重PCR反应模板,用优化后的反应条件进行双重PCR扩增,以检测其敏感性。
1.6 双重PCR反应条件优化建立10 μL反应体系:2×Taq DNA Master Mix 5 μL,2对特异性上、下游引物各0.5 μL,2种重组质粒标准品等比例混合物1 μL,ddH2O补足10 μL。对双重PCR引物浓度(终浓度为0.10,0.25,0.50,1.00 μmol/L 4个稀释度)、循环数(25,30,35,40个循环)及酶种类(Taq、LAMP、KOD)等反应条件进行优化。
1.7 双重PCR特异性试验采用建立的多重PCR方法,分别以重组质粒标准品等比例混合物和猫细小病毒(FPV)、猪伪狂犬病病毒(PRV)DNA或猪流行性腹泻病毒(PEDV)和猪传染性胃肠炎病毒(TGEV)cDNA为模板进行PCR扩增,评价该方法特异性。
2 结果
2.1 PPV5/7检测引物设计及系统发育分析如图1所示,在PPV5和PPV7的NS1基因分别找到2段高度保守序列,经对GC含量和退火温度优化后设计PPV5(285 F/R)和PPV7(630 F/R)2对引物(图1A),其中PPV5(285 F/R)这对引物片段与亲缘关系较近的PPV4相差较大,并且可扩增的285 bp 目的片段经Mega7构建Neighbor-Joining系统发育树(the sum of branch length=1.207 367 31,bootstrap=1 000)后能很好的与PPV4和PPV6区分开,聚类成单独的支系(图1B)。在PPV7(630 F/R)这对引物片段与亲缘关系较近的PPV6区别较大;同样的,通过对630 bp片段进行系统发育树(the sum of branch length=0.492 286 50,bootstrap=1 000)的构建后发现,该片段能够与PPV4、PPV5和PPV6区分开,聚成单独的支系,与WANG等[12]、JIN等[15]研究中,根据NS1基因构建的系统发育树树形一致[12,15],符合预期设定。

A.PPV5和PPV7基因组特点及双重检测引物设计示意图;B.PPV5引物保守性及285 bp目的片段系统发育树的构建;C.PPV7引物保守性及630 bp目的片段系统发育树的构建
2.2 重组质粒制备构建与验证挑选3个单克隆,提取质粒,以重组质粒pBM16A-PPV5和pBM16A-PPV7为模板利用表1中引物进行PCR扩增,pBM16A载体通用引物M13F/M13R作为阳性对照。结果显示,PPV5扩增出约为285 bp目的片段,PPV7扩增出约为630 bp目的片段,结果与预期大小一致(图2)。pBM16A-PPV5和pBM16A-PPV7测序结果与对应的PPV5和PPV7基因核苷酸序列一致。
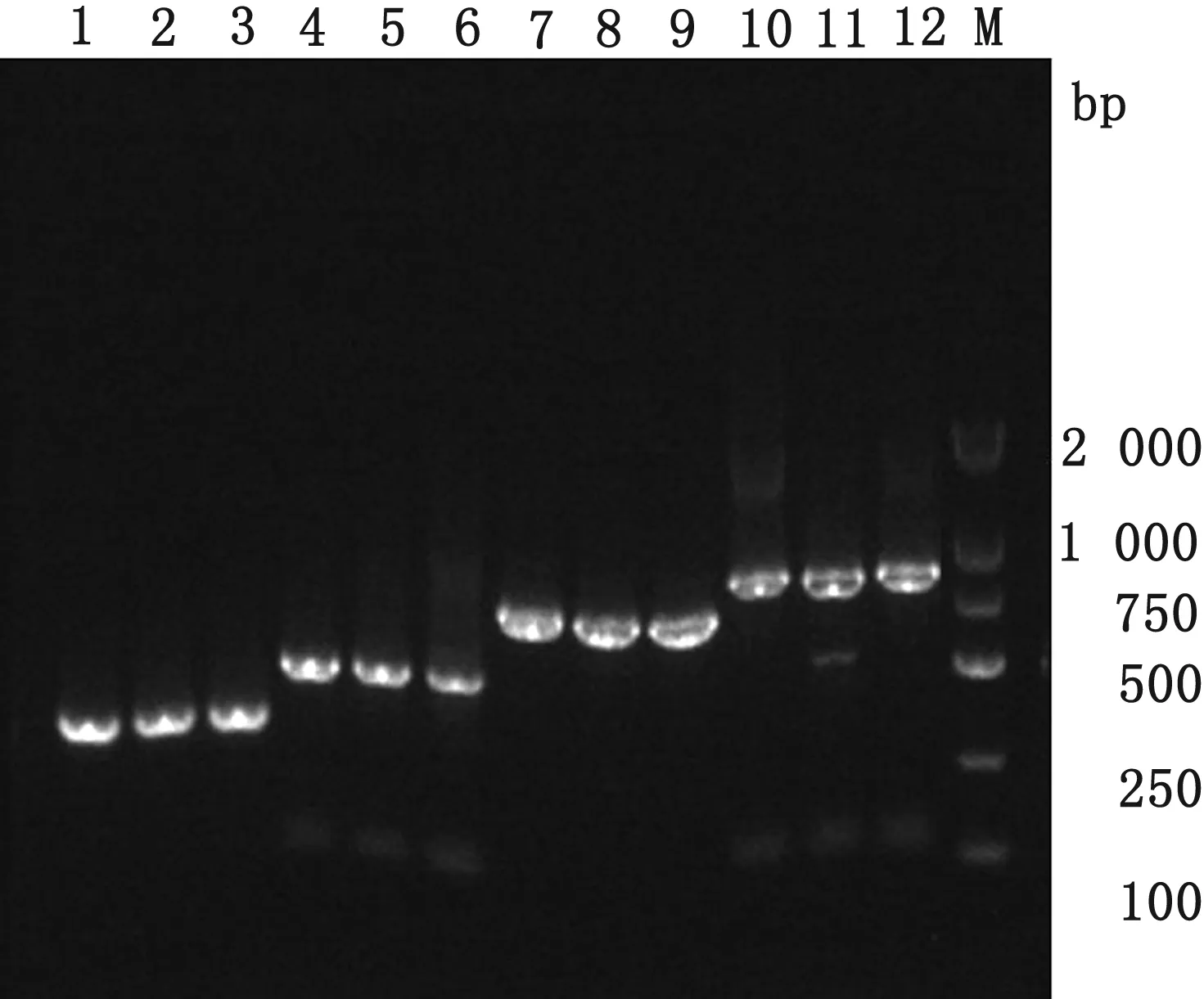
M.DL2000 DNA Marker;1~3.pBM16A-PPV5 PPV5F/R PCR扩增产物;4~6.pBM16A-PPV5阳性对照;7~9.pBM16A-PPV7 PPV7F/R PCR扩增产物;10~12.pBM16A-PPV7阳性对照
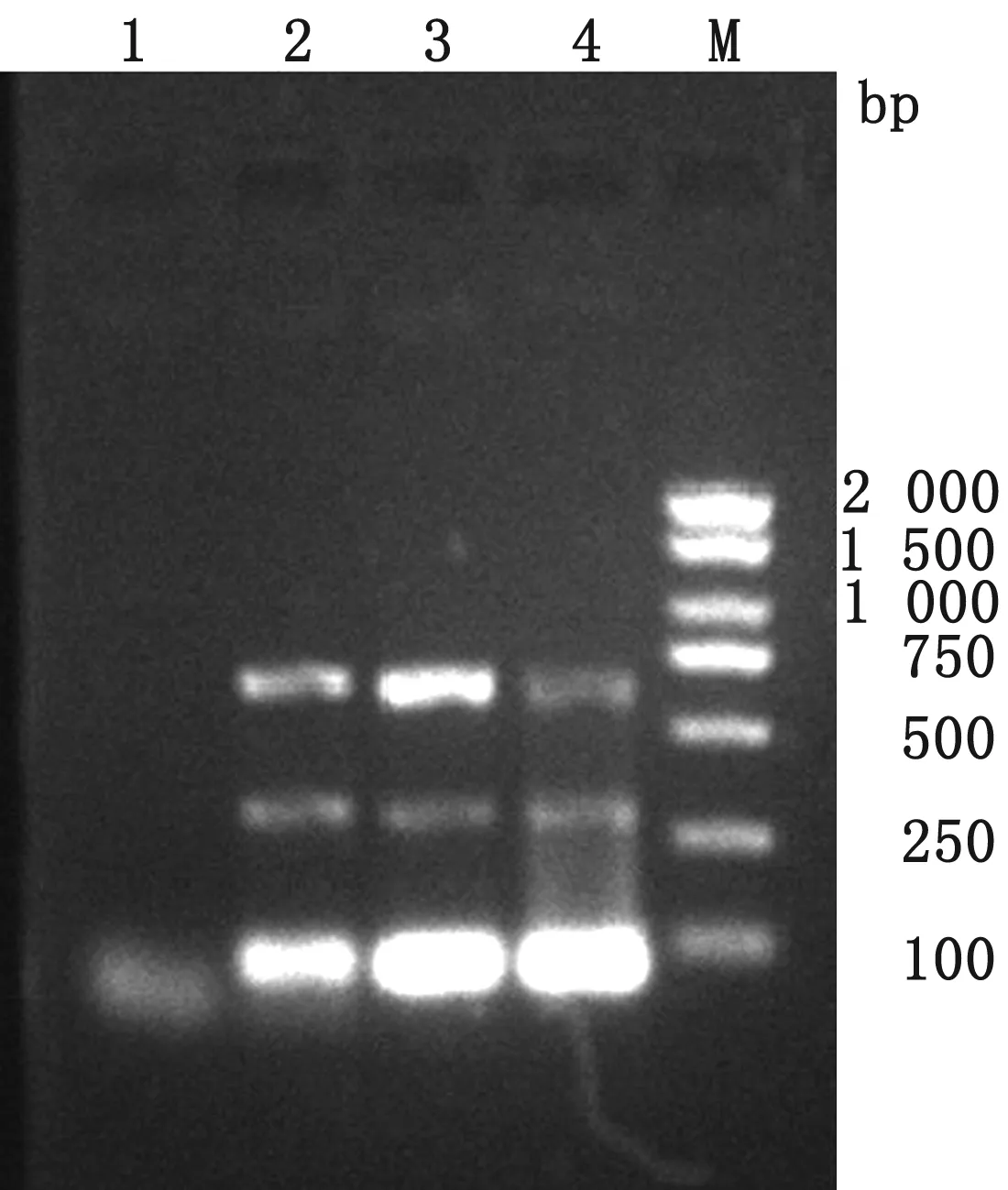
2.3 敏感性试验结果分别将2个重组质粒标准品10倍倍比稀释成终浓度为1.0×1012~1.0×101拷贝/μL,进行单一PCR反应。再取1.0×101~1.0×108拷贝/μL的2种重组质粒标准品并等体积混合进行双重PCR反应。结果显示,单一PCR检测PPV5下限为10拷贝/μL(图3A)、PPV7下限为10拷贝/μL(图3B),双重PCR检测这2种重组质粒标准品下限也为10拷贝/μL(图4),表明所建立的双重PCR敏感性较高。
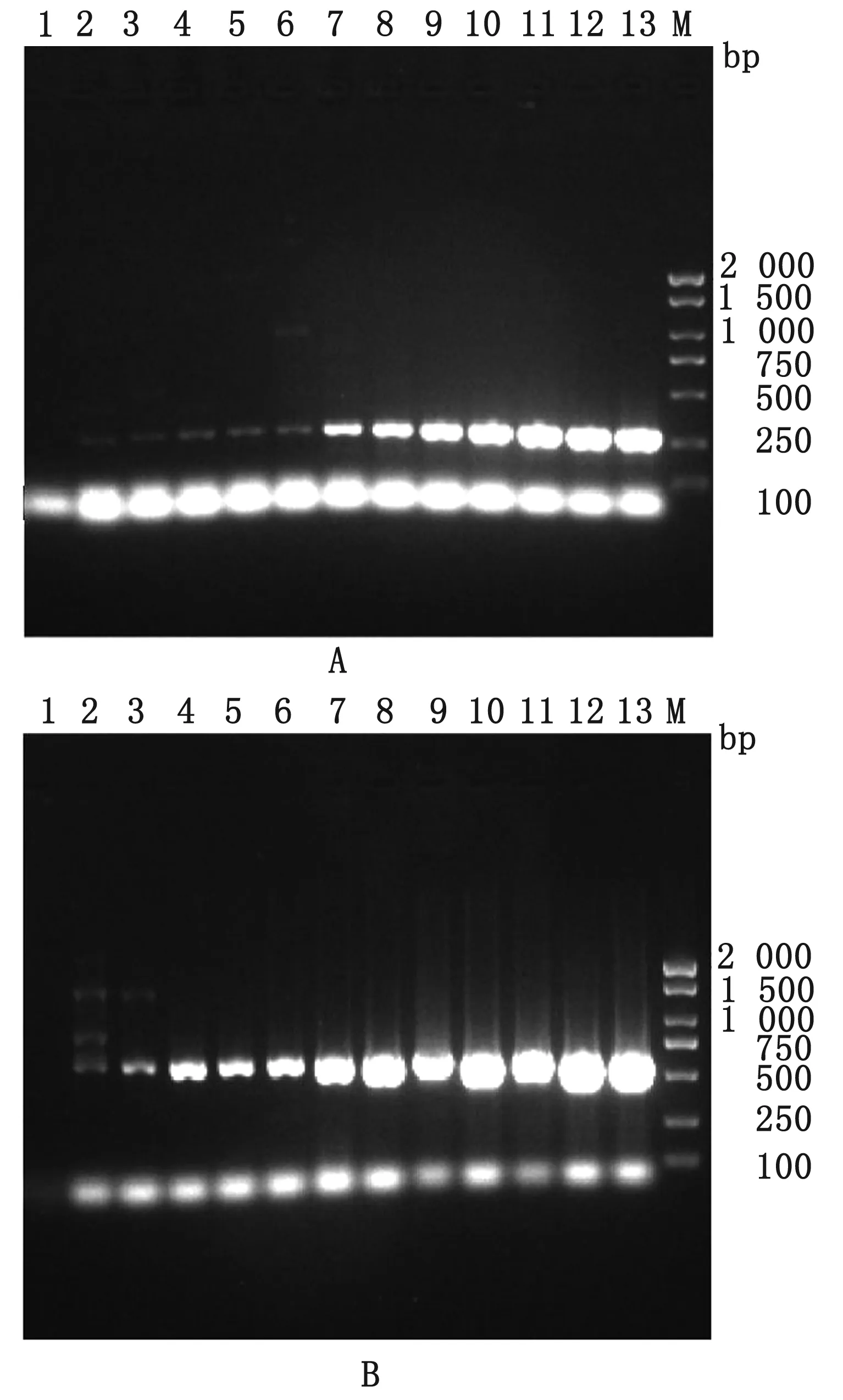
M.DL2000 DNA Marker;1.阴性对照;2~13.101~1012拷贝/μL PPV5(A)或PPV7(B)
M.DL2000 DNA Marker;1.阴性对照;2~9.PPV5和PPV7混合质粒101~108 拷贝/μL
2.4 双重PCR特异性试验结果应用建立的多重PCR方法,以不同病原DNA或cDNA为模板进行PCR扩增。结果显示,建立的多重PCR方法仅能扩增PPV5、PPV7基因组DNA,且与预期大小相符,而对FPV、PRV、PEDV、TGEV的核酸均无特异性扩增(图5),表明本研究建立的方法特异性较强。
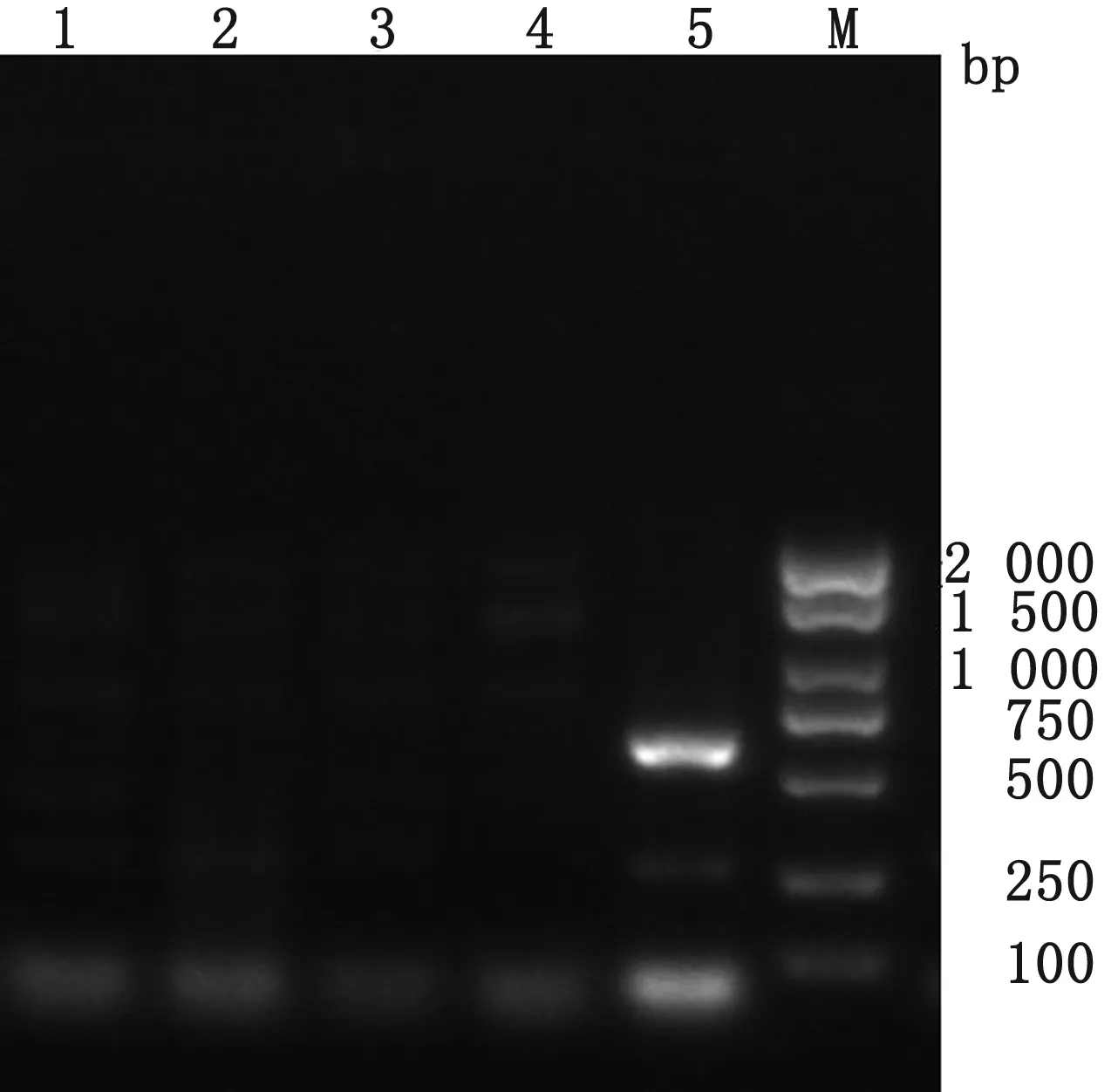
M.DL2000 DNA Marker;1.PRV;2.TGEV;3.PEDV;4.FPV;5.阳性对照
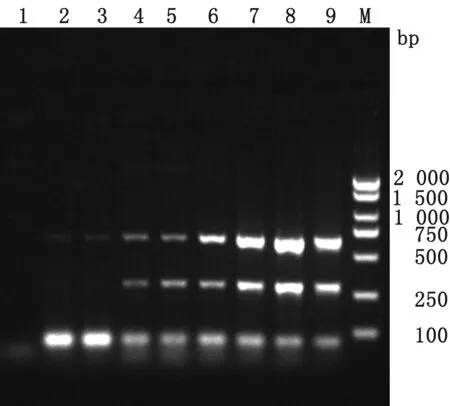
2.5 双重PCR方法条件优化结果选择浓度为1.0×105,1.0×106,1.0×107拷贝/μL的混和模板对反应条件进行优化,确定双重PCR反应的特异性上、下游引物最佳终浓度均为0.5 μmol/L(图6)。在1.0×103拷贝/μL浓度下对反应循环数进行优化,确定最佳扩增循环数为35个(图7)。选择1.0×105,1.0×106,1.0×107拷贝/μL对3种品牌的DNA聚合酶(LAMP、Mix、KOD酶)进行筛选,根据条带梯度、亮度和成本计算,认为2×Taq DNA Master Mix为该体系的最适检测用酶(图8)。
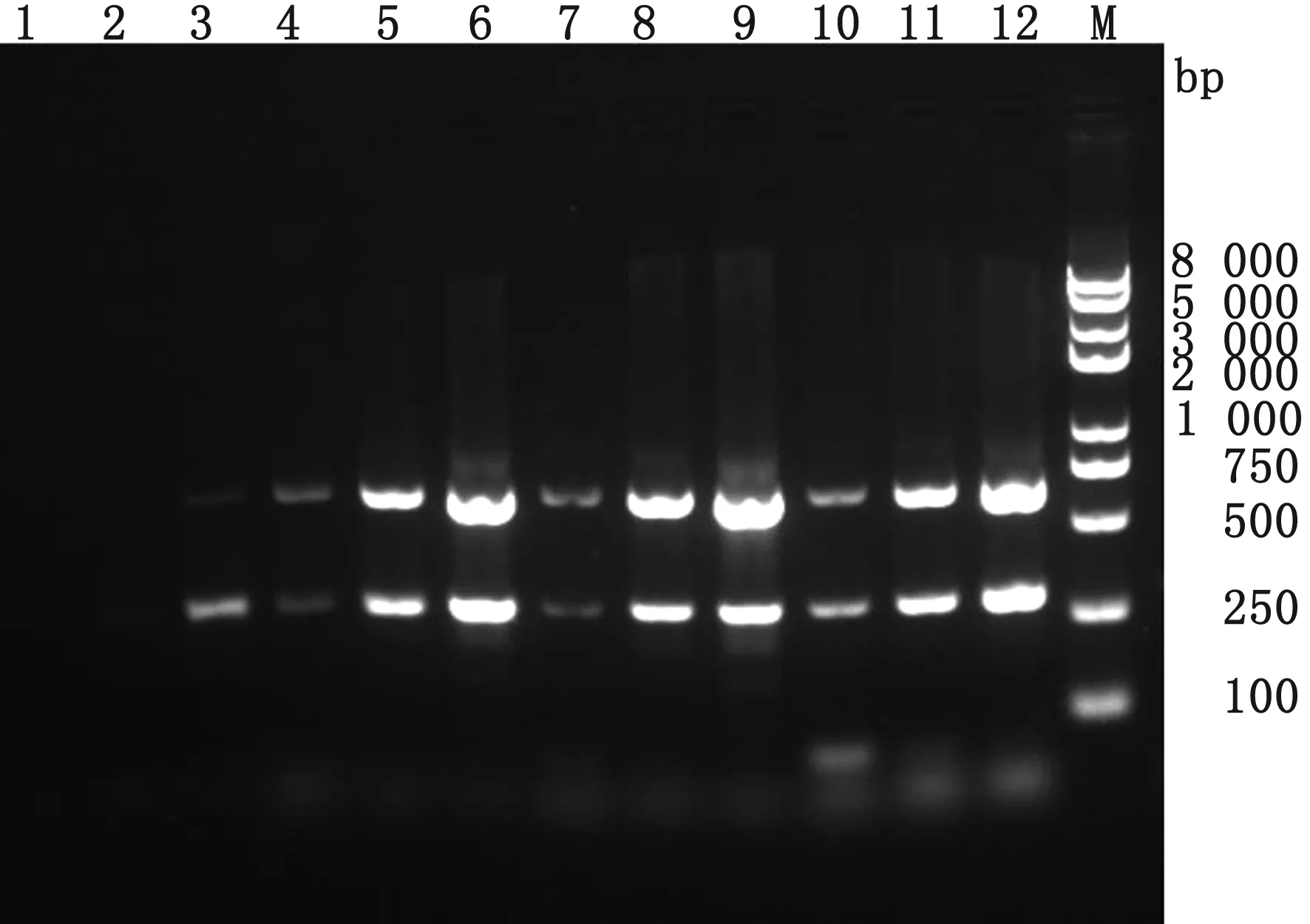
M.DL2000 DNA Marker;1~3.0.10 μmol/L;4~6.0.25 μmol/L;7~9.0.50 μmol/L;10~12.1.00 μmol/L
2.6 临床样品检测采用建立的多重PCR方法对黑龙江地区9只疑似患病猪的组织样品进行检测,其中样品1,2,5,8为肺组织样品,3,7为肠组织样品,4,6,9为脾组织样品。样品2,3,6,9和10均检测到285 bp目的条带,样品1,2,4和5检测出630 bp 目的条带,表明2,3,6和9号猪都感染PPV5,而1,2,4和5号猪感染PPV7,其中2号猪为PPV5和PPV7混合感染。
M.DL2000 DNA Marker;1~4.循环数为25,30,35,40

M1~M3.DL2000 DNA Marker;1~3.LAMP酶;4~6.Taq酶;7~9.KOD酶
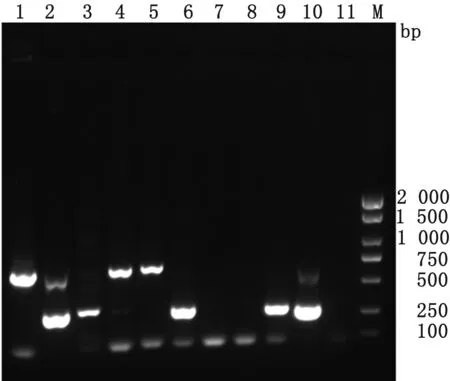
M.DL2000 DNA Marker;1~9.对应猪临床样品;10.阳性对照;11.阴性对照
3 讨论
近年来在我国贵州、湖南、广东、安徽和福建等地区均有PPV7、PPV5感染的病例[13]。PPV与PRV、猪呼吸与繁殖综合征病毒、猪圆环病毒2型和猪瘟病毒相比,其妊娠母猪临床症状不明显,不容易引起养殖户的注意,而此类病毒能与其他病原混合感染,严重的可导致猪只死亡或变成僵猪,为临床诊断带来困难[16]。目前主要诊断方法有:病毒分离、血凝抑制试验和乳胶凝集试验等,其中血凝抑制试验相对简便,但由于要制备豚鼠红细胞以及被检血清必须经高岭土或其他方法处理而显得麻烦;乳胶凝集试验灵敏度不高,尤其是对隐性感染动物往往漏检;病毒分离和鉴定结果准确可靠,但是该方法费时费力,并且需要一定的技术条件和设备;PCR及荧光定量PCR诊断具有灵敏度高,特异性强,快速、简便等优点已成为诊断PRV的研究热点[17]。本试验所建立的PPV5、PPV7双重PCR,能在1次反应中对这2种病原体引起的传染病进行核酸诊断且特异、敏感、快速、简便,利于防止该病蔓延扩散,减少经济损失,对猪场净化病原具有潜在的应用前景。
PPV病毒粒子无囊膜,直径约20 nm,基因组为单股DNA,大小4.0~6.3 kb,2个末端序列形成复杂的回文发夹结构,全基因组包含2~3个主要的开放阅读框架,由5′端UTR、非结构蛋白(NS1)、结构蛋白(Cap)和3′端UTR组成。PPV5的衣壳蛋白(VP1)中存在Ca2+结合环(YXGXG)和磷脂酶A2(PLA2)催化中心的HDXXY保守基序,包括D63,据报道这是PPV进入和感染的先决条件[4]。PPV具有高替换率(每年10-3~10-5替换/位点)进化,且属内的核苷酸替换速率也有不同的特点,PPV7的NS1和Cap蛋白基因的核苷酸突变率高于PPV1~PPV4,揭示PPV的遗传异质性,可能是病毒适应各种环境条件的有效方法[18]。
本研究根据PPV 2个亚型的保守基因设计不同长度目的片段的特异性引物,扩增到285(PPV5),630 bp(PPV7)2段目的基因,通过琼脂糖凝胶电泳直接判定扩增结果,更直观和实用,且对以上2片段进行系统发育分析时,可以准确聚集在同一支系上,利于降低结果假阳性,并开展初步的病毒溯源工作。经多个试验条件优化结果表明,25次循环时模板浓度较低时无条带,30~35次循环时有条带,条带清晰稳定。使用LAMP酶时目标条带微弱甚至无目标条带,虽然KOD酶扩增的条带清晰但其价格昂贵,综合这2个因素,使用2×Taq DNA Master Mix最为合适。经体系优化后,双重检测体系检测下线达到了单一检测的灵敏度,为10拷贝/μL,可应用猪群的一般检测工作。此外,本研究设计的引物可以特异性检测PPV5/7,不受其他猪源病毒(如PRV、TGEV和PEDV)的干扰,而且也无法检测到同属的猫细小病毒(FPV)。随机对2020年期间收集的黑龙江地区9头病猪的不同组织样品检测发现,存在1例共感染现象,PPV5和PPV7感染比例44.4%(4/9)。结果表明,本研究所开发的检测方法,具有临床应用价值,可在后续大群筛查中得以应用。
猜你喜欢
中国外汇(2019年7期)2019-07-13
智富时代(2019年2期)2019-04-18
智富时代(2019年2期)2019-04-18
中国生殖健康(2018年1期)2018-11-06
行政法论丛(2018年2期)2018-05-21
系统工程与电子技术(2016年2期)2016-04-16
计算机与网络(2015年12期)2015-06-21
中国光学(2015年1期)2015-06-06
海岸工程(2014年4期)2014-02-27
中国科技信息(2011年12期)2011-02-17
