
超高效液相色谱-串联质谱法测定鹅可食用组织中维吉尼亚霉素M1的残留量
2021-11-04 09:59王霄旸李继东薛飞群张可煜王春梅
中国动物传染病学报 2021年5期
高 蕊,王霄旸,李继东,薛飞群,张可煜,王春梅,周 文
(1.宁夏大学农学院,银川 750021;2.中国农业科学院上海兽医研究所,上海 200241)
维吉尼亚霉素(virginiamycin, VGM)又名纯霉素、维及霉素、威里霉素、抗金葡霉素等,是由维吉尼亚链霉菌发酵产生的链阳性菌素。这类抗生素中的每一个成员至少由两种结构不相关的分子组成,维吉尼亚霉素则主要由M1与S1两种抗菌因子构成,M1因子为大环内酯,S1因子为环状多肽。结构式见图1。当M1与S1因子的比例构成达到7∶3时,其抗菌活性最强。维吉尼亚霉素通过阻止细菌蛋白质的合成而达到杀菌作用[1],主要对八叠球菌、枯草杆菌、金黄色葡萄球菌等革兰氏阳性菌有抑制作用,可用于治疗肝脓肿[2]、鸡坏死性肠炎[3]等疾病。同时该药具有明显的促生长作用,能提高饲料的利用率[4-5],也因此作为饲料添加剂在畜禽中广泛使用。

图1 维吉尼亚霉素M1(A)和维吉尼亚霉素S1(B)Fig.1 Virginiamycin M1 (A) and Virginiamycin S1 (B)
经过研究表明,维吉尼亚霉素的使用会降低某些药物的药效,例如普那霉素(Pristinamycin)[6]。一些学者研究发现维吉尼亚霉素的持续使用可能会通过粪便传播、机体残留蓄积等方式增加人类对链球菌耐药的屎肠球菌感染的可能性[7-8],维吉尼霉素发挥作用的主要是M1组分,且在机体内代谢比较缓慢。因此检测维吉尼亚霉素残留时常将维吉尼亚霉素M1组分作为残留标识物。加拿大作为维吉尼亚霉素预混剂的原研国,已经将维吉尼亚霉素残留标识物修订为M1,并在“食品中兽药最大残留限量(maximum residue limit, MRL)清单中规定,鸡和猪的肝脏、皮脂、肌肉、肾脏的MRL分别为300 μg/kg、400 μg/kg、100 μg/kg和400 μg/kg”。我国农业农村部2019年发布的GB 31650-2019《食品中兽药最大残留限量》,也将残留标示物修订为M1,其中规定了家禽和猪的肌肉、皮脂、肝、肾的MRL分别为100 μg/kg、400 μg/kg、300 μg/kg和400 μg/kg,出于交叉耐药性的考虑,欧盟于1999年起禁止维吉尼亚霉素作为动物的饲料添加剂使用,而从2006年1月起,更是全面禁止食品动物使用抗生素作为促生长饲料添加剂。我国也自2021年7月1日起规定,包括维吉尼亚霉素预混剂在内的12种促生长药物饲料添加剂(AGPs)被禁止生产、进口、经营和使用。
此前已经建立了检测猪、鸡可食性组织中维吉尼亚霉素M1残留的检测方法[9],很少有人去关注鹅可食性组织中维吉尼亚霉素M1的残留情况。俄罗斯、中亚各国、中东伊斯兰教国家都喜食鹅肉,国际上对鹅肉、鹅肝等需求量呈明显的增长趋势。鹅作为常见家禽在我国养殖中也占了很大的比重。因此,建立一种快捷、高效的检测鹅可食性组织中维吉尼亚霉素M1残留量的方法是必要的,不仅提高了检测效率,也为建立标准检测方法奠定基础。
1 材料与方法
1.1 仪器设备 超高效液相色谱仪Waters Acquity UPLC system、电喷雾质谱仪Waters TQ-S购自美国Waters公司;分析天平XS105DR购自梅特勒托利多公司;精密天平XS4002S购自梅特勒托利多公司;超声波清洗仪KQ-500DE购自上海超声波仪器厂;离心机LXJ-IIB购自上海安亭科学仪器厂;涡旋混合器Genie-2购自Scientific Industries公司;高速匀浆机购自Waring公司。
1.2 材料试剂 空白鹅购自杭州萧山汉潮家禽有限公司;维吉尼亚霉素M1对照品,CAS Number:21411-53-0,购自Sigma公司,美国,纯度:含量≥93%。乙腈为色谱纯,购自塞默飞世尔公司;甲酸为色谱纯,购自Sigma公司;正己烷为分析纯,购自国药试剂化学品有限公司。
1.3 标准溶液的配制 准确称取维吉尼亚霉素M1对照品5.0 mg,置于100 mL棕色容量瓶中,用适量乙腈使其溶解并稀释至刻度,配制成浓度为50 μg/mL的维吉尼亚霉素M1标准贮备液。维吉尼亚霉素M1标准中间液:准确量取维吉尼亚霉素M1标准贮备液,并用乙腈稀释至刻度,配制成浓度为0.1、0.3、0.5、1、1.5、2、3、4、6、8和9 μg/mL的维吉尼亚霉素M1标准中间液。标准工作液:使用前将维吉尼亚霉素M1标准中间液用乙腈-水(60∶40)混合液稀释成浓度为2、10、20、40和60 ng/mL的维吉尼亚霉素M1系列标准工作液,供液相色谱联用质谱仪测定。
1.4 质谱条件的优化 采用Infusion方式对维吉尼亚霉素M1直接进行质谱方法摸索,扫描范围设定为m/z 80~600,结果可见M1的分子离子峰[M+H]+526.3。在正离子扫描模式下,进行多反应监测方法筛选子离子,得到355.1和337.1两个响应较高的子离子,优化碰撞能(collision energe,CE),质谱条件为:离子源温度为150℃,脱溶剂温度为200℃,毛细管电压为3.0 kV,脱溶剂气流速为800 L/h,锥孔反吹气流速为150 L/h,定性、定量离子对、碰撞能量和锥孔电压,见表1。维吉尼亚霉素M1的全扫描质谱图,见图2,维吉尼亚霉素M1子离子扫描图,见图3。

表1 维吉尼亚霉素M1的定性、定量离子对、碰撞能量和锥孔电压Table 1 The qualitative and quantitative ion pair of VGM-M1 and the collision energy and cone voltage
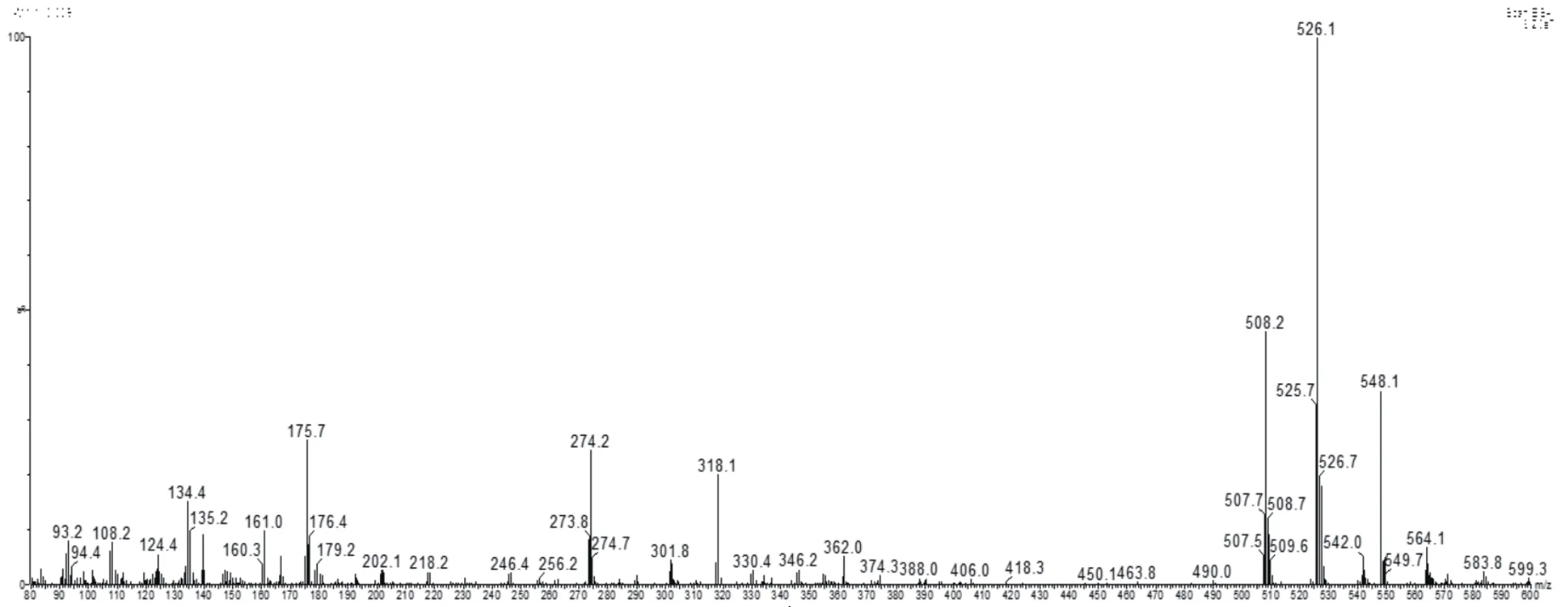
图2 维吉尼亚霉素M1全扫描图(碰撞能量:4eV)Fig.2 Full scan of VGM-M1 (collision energy: 4eV)

图3 维吉尼亚霉素M1子离子扫描图(碰撞能量:18eV)Fig.3 Scanning image of VGM-M1 daughter ion (collision energy: 18eV)
1.5 液相条件的优化 采用了Waters Atlantis UPLC BEH C18 液相色谱柱(1.7 μm,2.1×100 mm),流动相A为乙腈,流动相B为0.1%甲酸水,柱温:30℃;进样量:5 μL;流速:0.2 mL/min;经过液相条件的优化,最终采用梯度洗脱的方式。梯度洗脱程序为:初始30%A;0~1 min,A由线性升至30%;3~6 min,A线性升至42%;6.5~8.5 min,A线性升至95%;9~10 min,A线性降为30%。保留时间为5.27 min。
1.6 样品前处理过程的优化 称取均质组织2.00 g(±0.02 g),置于50 mL离心管中,其中肌肉组织加入2 mL水,涡旋2 min,加入4 mL乙腈;其他组织直接加入4 mL乙腈,各组织加入乙腈后涡旋2 min,超声30 min, 4000×g离心10 min,将上清液转移至15 mL具盖离心管中。再次向残渣中加入2 mL乙腈,重复提取一次,合并上清液,备用。在合并的提取液中,加水至约9.5 mL,涡旋混合30 s,加入3 mL正己烷除脂,涡旋30 s,4000×g离心10 min,弃去上层正己烷层,重复除脂一次,尽量完全弃去上层正己烷后,转移至10 mL刻度试管中,加水至10 mL,涡旋混合30 s,备用。
肌肉试料经上述过程处理后,提取液0.22 μm滤膜过滤,取续滤液,供液相色谱联用质谱仪测定。肝脏试料处理后准确量取2.0 mL提取液溶液加入4.0 mL乙腈-水(60∶40)混合液,涡旋30 s混合后,0.22 μm滤膜过滤,取续滤液,供液相色谱联用质谱仪测定。肾脏和皮脂试料处理后分别准确量取2.0 mL提取溶液加入6.0 mL乙腈-水(60∶40)混合液,涡旋30 s后,0.22 μm滤膜过滤,取续滤液,供液相色谱联用质谱仪测定。
1.7 方法学研究
1.7.1 标准溶液稳定性研究 取新鲜配制的维吉尼亚霉素M1贮备液,稀释成0.02、0.06和0.08 μg/mL的标准工作液,分别存贮在-20℃、4℃、室温避光及20℃光照条件下,同一条件下每个水平有两个平行样,计算3个浓度的含量百分比。
1.7.2 基质效应研究 按1.3配制系列维吉尼亚霉素M1标准工作液,浓度为2、10、20、40和60 ng/mL,进样测定后绘制标准曲线A。取各个组织的空白试样,按照1.6方法处理空白样品,在定容前加入相应浓度的维吉尼亚霉素M1标准中间液,再定容或稀释,0.22 μm滤膜过滤,进样测定后,绘制各个组织维吉尼亚霉素M1系列浓度的基质添加标准曲线B;通过计算标准曲线B与标准曲线A的斜率比值判断是否有基质效应。
1.7.3 特异性 按照1.6样品前处理方法对空白试样和空白添加试样提取制备后,按照1.4和1.5的液相、质谱方法进行进样测定,观察是否有空白组织是否对维吉尼亚霉素M1存在干扰。
1.7.4 线性 按照1.3的方法配制成维吉尼亚霉素M1浓度为2、10、20、40和60 ng/mL系列标准工作液,鹅的肌肉的基质添加标准工作曲线,供液相色谱-串联质谱仪测定。以待测物的定量特征离子质量色谱峰面积值为纵坐标,待测物标准溶液的浓度为横坐标绘制标准曲线,得到线性方程。
1.7.5 灵敏度 空白添加试样按照1.6处理后,按照1.4和1.5中的液相、质谱条件进行检测,以待测物的信噪比S/N≥3为方法的检测限,信噪比S/N≥10为方法的定量限为原则,确定组织中维吉尼亚霉素M1的检测限和定量限。
1.7.6 精密度和准确度 采用标准添加法,在空白试料中添加定量限浓度、MRL的0.5倍MRL、1倍MRL、2倍MRL的4个水平添加浓度来进行精密度和回收率的考察。每个浓度5个平行样品,3个批次。以测定浓度与添加浓度的百分比计算试样的百分回收率;以批内5个样品百分回收率之间的RSD%计算批内精密度;以3个批次15个样品百分回收率之间的RSD%计算批间精密度。
2 结果
2.1 标准溶液稳定性研究 标准溶液稳定性测定结果见表2,结果表明,标准溶液在4℃条件、室温避光条件下放置30 d后含量在94.46%-107.90%,稳定性较高,在-20℃、室温光照条件下,10 d以后测定值波动较大,为保证检测的准确,因此将标准溶液保存时间为4℃避光保存7 d。
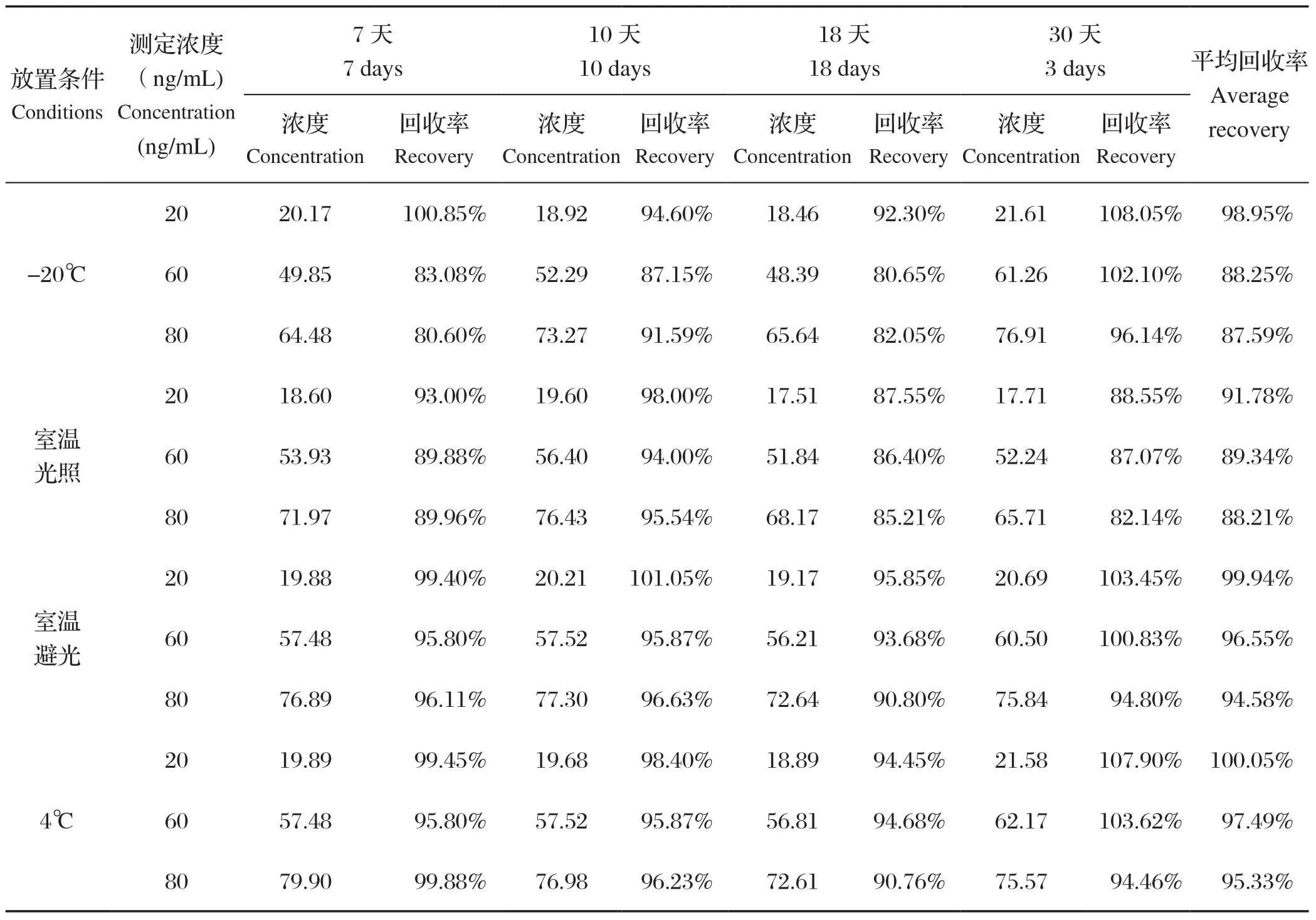
表2 标准溶液稳定性结果(n=2)Table 2 Stability results of standard solution (n=2)
2.2 基质效应研究 一般规定,斜率比值在85%~115%时认为不存在基质效应。结果显示,鹅的肌肉组织标准曲线B与标准曲线A的斜率比值为171.5%,存在基质增强效应,其他组织的斜率比值均在85%~115%范围内,因此在测定样品时鹅的肌肉组织采用基质添加工作曲线,其他组织采用标准工作曲线,见表3。

表3 鹅可食性组织中维吉尼亚M1基质效应Table 3 Matrix effect of VGM-M1 in edible tissue of geese
2.3 特异性 空白试样和空白添加试样的色谱图,见图4。结果显示,鹅可食性组织基质对维吉尼亚霉素M1的检测无干扰,峰型良好,出峰时间适宜。
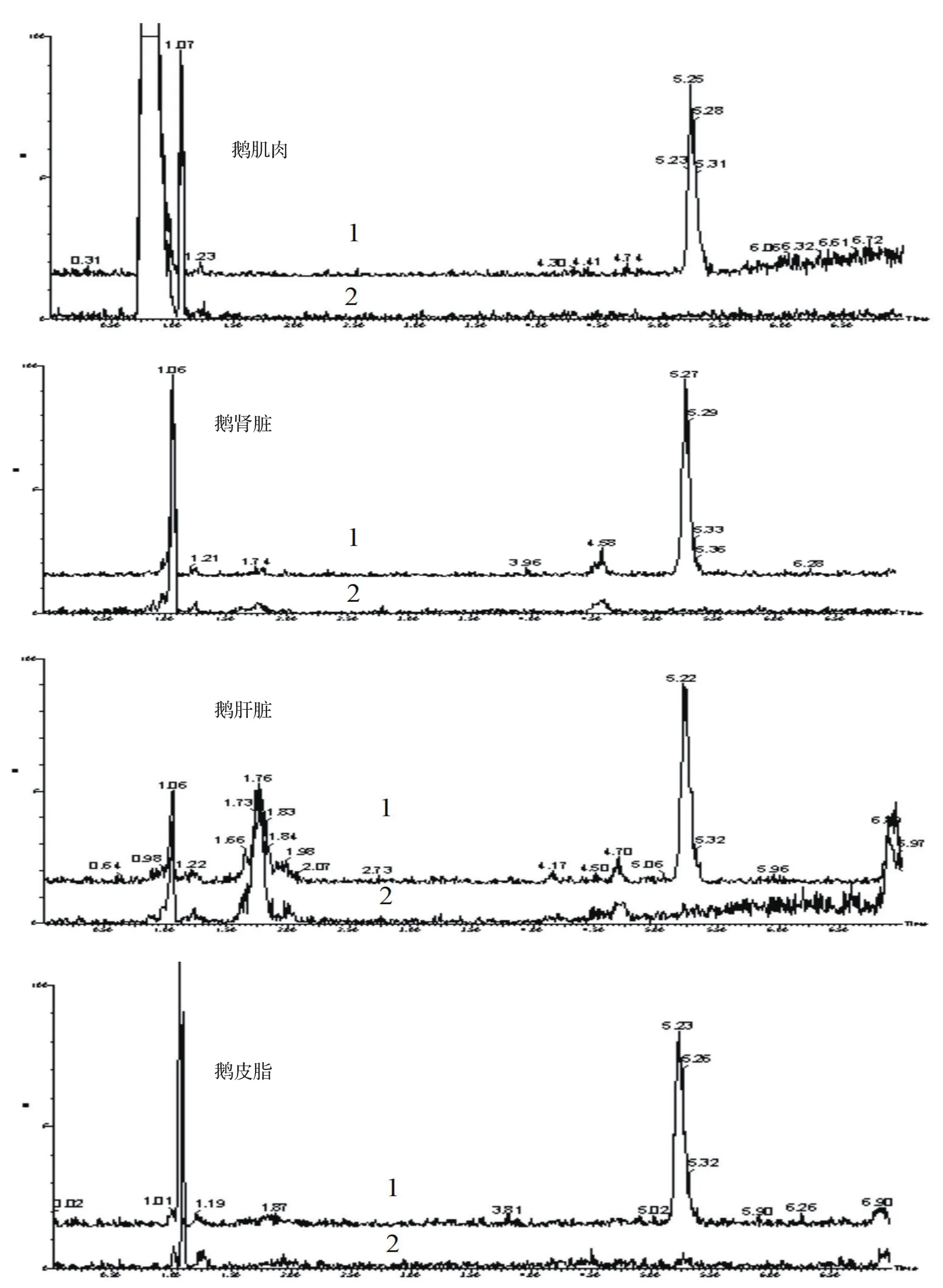
图4 鹅可食性组织中维吉尼亚霉素M1的色谱图Fig.4 Chromatogram of VGM-M1 in edible tissue of geese
2.4 线性 以待测物的定量特征离子质量色谱峰面积值为纵坐标,待测物标准溶液的浓度为横坐标绘制标准曲线,得到线性方程,R2均大于0.99,维吉尼亚霉素M1在2.011~60.68 ng/mL检测浓度范围内,呈良好的线性关系(权重系数为1/x)。
2.5 灵敏度 以待测物的信噪比S/N≥3为方法的检测限,信噪比S/N≥10为方法的定量限为原则,测得鹅可食性组织中维吉尼亚霉素M1的检测限和定量限如表4所示。此方法的灵敏度可满足维吉尼亚霉素M1的检测技术指标要求。
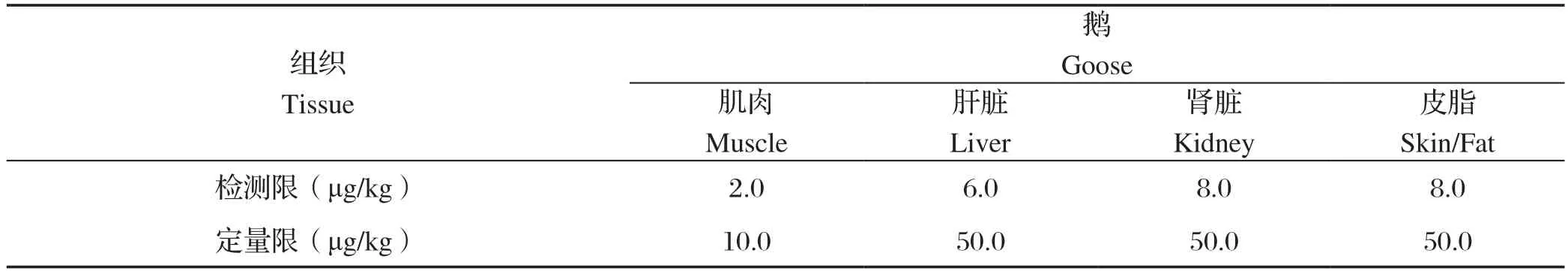
表4 维吉尼亚霉素M1的检测限和定量限Table 4 Limit of detection and limit of quantification of VGM-M1
2.6 精密度和准确度 根据NY/T 1896-2010“兽药残留实验室质量控制规范”中针对准确度和精密度试验的规定将定量限、0.5MRL、MRL、2MRL设定为添加浓度。GB 31650-2019《食品中兽药最大残留限量》中维吉尼亚霉素M1的最大残留限量值见表5。由表6可知,在各个浓度下,本方法的回收率、批内变异系数和批间变异系数均符合兽药残留规范的相关要求,具有较好的回收率和精密度。

表5 维吉尼亚霉素M1在各个组织中MRLs值Table 5 The MRLs value of VGM-M1 in various tissues
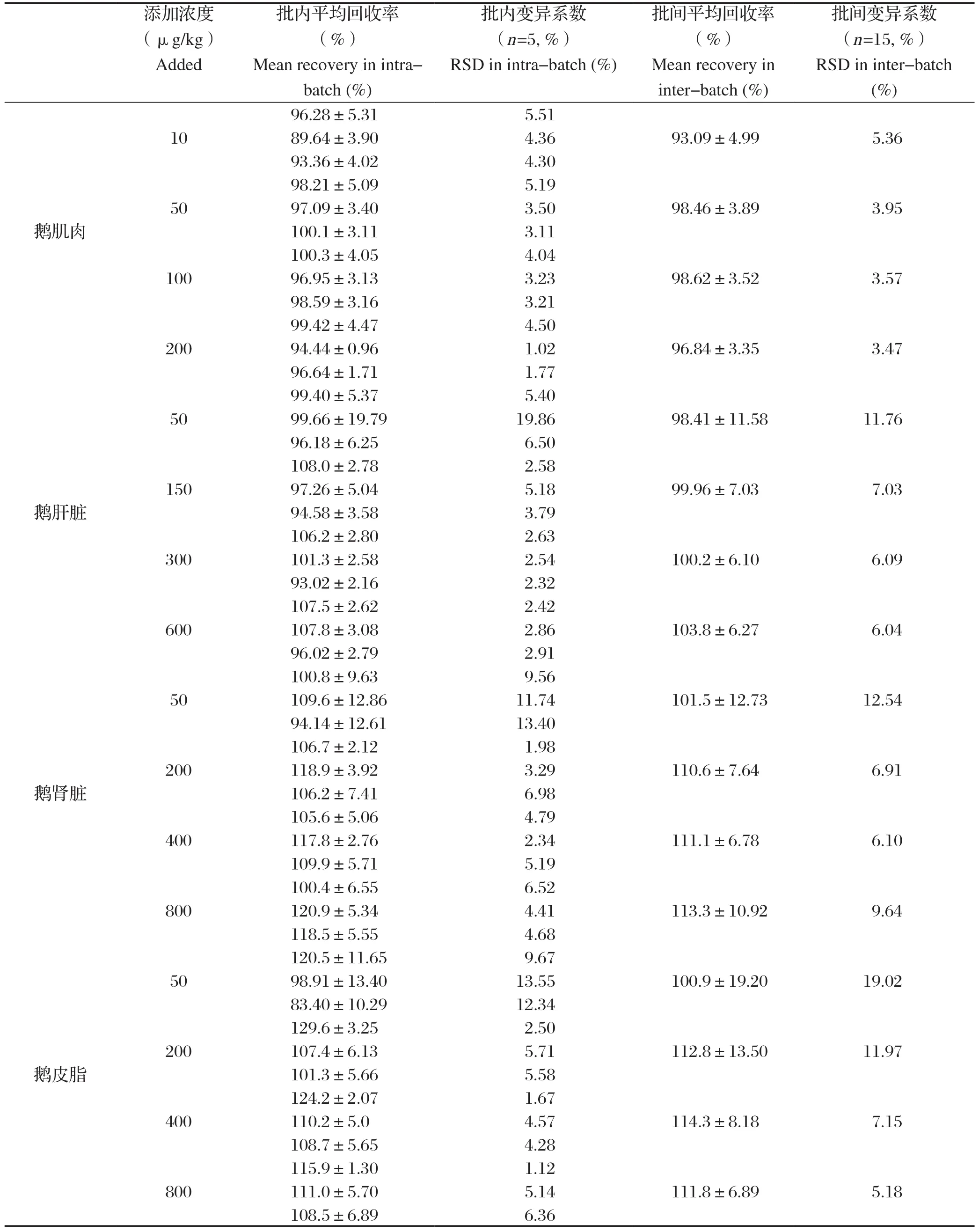
表6 维吉尼亚霉素M1在鹅各组织中的添加回收率Table 6 The spiked recoveries of VGM-M1 in goose organizations
3 讨论
目前采用的组织中维吉尼亚霉素残留检测方法有 微生物法、酶联免疫法、液相色谱法以及液相色谱-质谱联用法等。微生物法和酶联免疫法检测结果受很多因素的影响,导致检测结果出现重复性差、准确度不高、检测线较高等缺点,难以实现标准化,检测结果也需要进行确证试验排除假阳性结果,存在很多局限性[10]。高效液相色谱法、液相色谱-质谱联用法具有较高的准确度、灵敏度、更快的分析速度越来越多的应用于残留检测[11]。本试验采用超高效液相色谱-串联质谱法检测,分辨率和灵敏度高,基体干扰少,检测样本数量大,分离速度更快,缩短了分析时间。
不同的样品前处理技术对样品检测也有影响,快速、有效的提取方法不仅提高检测速度,也提高了检测效率。Boison等[12]建立了运用LC-MS测定猪的肝脏、肾脏和肌肉组织中维吉尼亚霉素M1残留量。组织样品用10 mL甲醇-乙腈(1∶1)匀浆,经两次提取后离心,取其上清液,加入15 mL 0.01 mol/L磷酸铵溶液,用C18固相萃取柱净化,用水-甲醇(45∶55)洗脱到10 mL氯仿中,去除水层,吹氮气将氯仿蒸发。用甲醇和甲酸氨缓冲液溶解并定容,通过0.22 mol/L圆盘过滤器过滤,进行LC-MS分析,定量限为2 mg/kg。该试验处理样品过程中应用到有毒溶剂氯仿,而且定量限较高,并不适合推广。杜业刚等[13]运用UPLC-MS/MS法同时测定动物源性食品(鸡肉、鸭肉、猪肉、牛肉、羊肉、猪肝、猪肾、鸡肝、鸡肾、鱼)中8种多肽类抗生素,样品经甲醇-水-甲酸(40∶60∶0.1)、1 mol/L硫酸溶液提取,正己烷进行脱脂,HLB固相萃取柱净化,甲醇-水-甲酸(70∶30∶0.1)洗脱,乙腈-水-甲酸溶液(10∶90∶0.1)溶解定容,过滤后,进行测定。以0.1%甲酸水溶液和0.1%甲酸乙腈溶液为流动相,选择反应监测(selected reaction monitoring,SRM),外标法定量。结果表明,维吉尼霉素M1检出限为0.5 μg/kg,维吉尼霉素S1检出限为0.1 μg/kg,定量限均为1 μg/kg。回收率为61.0%~78.2%。这种方法在样品处理方面选用的试剂较多,可能是导致回收率不高的原因之一。
本试验中对提取剂进行了筛选,选出了提取效率高的提取剂。根据维吉尼亚霉素的理化性质,查阅相关文献和标准后,分别考察了采用乙腈、甲醇、乙酸乙酯及甲醇乙腈按1∶1混合溶液对样品进行提取的回收率,结果发现,采用乙腈的提取效率最高,一次提取回收率就能达到70%以上,两次提取后,回收率均能达到90%以上,所以在后续试验操作中选择乙腈作为提取剂。由于组织样品中脂肪含量较高,为避免影响后续实验操作需对样品进行除脂,本试验中采用正己烷作为除脂剂,将除脂后的正己烷层进行吹干再用流动相溶解后,进行检测,未发现有M1,表明使用正己烷除脂不会干扰M1的检测,不影响M1的提取效率。超高效液相-质谱联用灵敏度较高,在测定标准溶液时,最低检测限可以达到进样量10.0 pg,而维吉尼亚霉素在各个组织中的最高残留限量在100~400 μg/kg,远远高于仪器的检测限,因此在建立方法时,考虑到样品前处理方法的简便快速,取消了样品浓缩步骤,减少了前处理时间,也降低了吹干溶剂时对样品稳定性的影响。标准溶液的配制溶剂对液相峰型有较大影响,在对维吉尼亚霉素M1液相分离条件进行探索时发现,只有在乙腈-水比例接近6∶4时才能得到较好的峰型,因此在乙腈提取液中加入超纯水,调节进样液中溶剂比例。在前处理时,由于肌肉组织质地较硬,加入乙腈涡旋处理时,易结块,影响提取效率,故在取样后,先加入2 mL水,进行涡旋,再用乙腈进行提取,则不会出现明显的结块现象。由于每个组织的MRLs值不一样,而考虑到检测时标准曲线的线性范围要适合于所有组织的测定,并且满足于定量限回收率的检测要求,因此将肾脏、肝脏和皮脂组织在定容后按比例稀释,使其实际检测溶液浓度范围与肌肉组织一致。
本研究采用超高效液相色谱质谱联用法检测鹅可食性组织中维吉尼亚霉素M1的残留量,经过样品前处理过程的优化后,简化了提取过程,提高了提取效率。最终得到鹅各组织的定量限分别为:肌肉10.0 μg/kg、肝脏50.0 μg/kg、肾脏50.0 μg/kg、皮脂50.0 μg/kg,检测限均小于10 μg/kg。本研究建立的检测方法灵敏度高、准确度好、样品处理过程简便快速,通过灵敏度、线性、回收率、精密度等方法学验证后,各项指标均满足标准要求,因此本方法完全可满足我国市场对鹅可食性组织中维吉尼亚霉素M1残留量的检测要求。
猜你喜欢
煤化工(2022年3期)2022-07-08
口腔护理用品工业(2021年4期)2021-11-02
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
色谱(2021年7期)2021-06-07
雪豆月读·中年级(2020年12期)2020-09-10
祖国(2017年9期)2017-06-15
中国资源综合利用(2016年10期)2016-01-22
河北工业科技(2015年4期)2015-02-27
天津化工(2010年5期)2010-09-18
