
金属调控的2,4,6⁃三(2⁃吡啶基)⁃1,3,5⁃三嗪基配合物的光致变色性能
2022-02-17 07:37郝朋飞付云龙
无机化学学报 2022年2期
郝朋飞 刘 兴 付云龙
(山西师范大学化学与材料科学学院,磁性分子与磁信息材料教育部重点实验室,太原 030006)
0 引言
由电子给体−受体单元组成的有机−无机杂化光致变色材料由于灵活多样的组成和结构及良好的视觉可检测性的变色行为,在传感、保护、显示和开关等领域展现出重要的实际或潜在应用价值[1-5]。分子间电荷转移(CT)和电子转移(ET)是有机−无机杂化电子给体−受体系内部最基本的2种电子行为,已经成为决定材料刺激响应变色性能的关键因素。分子间的CT是杂化材料内部电荷的跃迁行为,决定着电子给体−受体杂化体系的起始发色和光响应范围,而分子间ET则是材料内部的电荷分离,决定着电子给体−受体杂化体系在光刺激下形成自由基的发色和稳定性以及后续的可逆性和循环性。由于所谓的重原子效应或极化效应,电子给体和受体之间较强的CT会引起电子发生快速重组,在一定程度上不利于光诱导分子间ET的发生,从而降低了杂化体系的光响应速率和变色前后色差。电子给体和受体之间较弱的CT虽然有利于形成变色前后明显的色差,但是会导致杂化体系内部电荷跃迁需要更高的能垒,从而使得杂化材料的激发需要更高的能量,在一定程度上限制了杂化材料的光响应范围。因此,分子间的CT和ET之间的关系既对立又统一,如何实现二者的微妙平衡是赋予电子给体−受体杂化体系明显色差、良好响应速率和优异可逆变色性能的关键科学问题。
最新研究表明,有效调控电子给体−受体的电子给−受能力匹配和界面关系匹配,是优化电子给体−受体杂化体系内部电子行为和光致变色性能的2个重要策略。值得注意的是,通过有效调控电子给体−受体的电子给−受能力匹配,将电子给体和受体单元通过特定方式进行自组装,在提高功能材料光响应速率、颜色对比度及相关性能方面已经取得了一些重要突破。2007年,郭国聪团队首次将具有优异缺电子特性的4,4′-联吡啶引入到氯铋酸盐体系,得到了一例基于分子间ET光致变色机制的晶态紫精−氯铋酸盐电子给体−受体杂化体系[6]。在此基础上,国内外多个研究小组对4,4′-联吡啶基电子给体−受体杂化变色材料开展了较为全面的研究,将缺电子受体4,4′−联吡啶及其衍生物,通过特定的方式分别与氯金属酸盐、羧酸盐、分子筛、磷酸盐以及含羧基配体/溶剂等电子给体进行杂化,构筑了大量具有优异光致变色性能的多功能杂化晶态材料,初步探明了4,4′−联吡啶及其衍生物缺电子受体与多种富电子给体在电子给−受能力之间的匹配规则[7-20]。例如,吉林大学于吉红院士和李激扬教授团队以原位生成的甲基紫罗碱为模板,指导合成了一系列具有多重外界刺激(包括可见光、紫外光、X射线和热)响应变色的分子筛材料[13-14];北京理工大学张杰教授课题组发展了一系列具有优异光致变色特性的紫罗碱基羧酸盐配合物[15-16];华东师范大学高恩庆教授课题组开发了基于分子间ET的压致变色、水致变色和光致变色的紫罗碱基金属有机框架材料[17-18];郑州大学臧双全教授团队将紫罗碱组分嵌入到新型稀土金属有机框架中,首次实现了光致变色、光调变发光和荧光pH传感的多重光功能调控[19-20]。为了进一步探索分子间的电子给−受能力匹配行为,国内外科研工作者在Saha等[21]关于溶液体系中1,4,5,8-萘二酰亚胺及其衍生物(NDIs)与各种阴离子之间CT和ET行为的研究基础上,将缺电子功能基团NDIs引入到多种富电子体系,成功构筑了一系列具有优异光致变色特性的NDIs基晶态杂化材料[22-25]。2011年,华南理工大学傅志勇教授团队将刚性的多吡啶基缺电子受体2,4,6-三(4-吡啶基)-1,3,5-三嗪(TPT)与羧酸盐电子给体结合,构筑了2个具有优异光致变色性能的金属有机框架,并详尽探讨了电子给体−受体杂化体系结构与光响应活性之间的关系[26]。随后,该研究团队又将缺电子组分TPT嫁接到无机金属磷酸盐−亚磷酸盐体系,构筑了一例有趣的光致发光和光致变色双功能电子给体−受体杂化体系,进一步拓宽了TPT缺电子受体与含氧电子给体的匹配范围[27]。2018年起,青岛大学王国明课题组将TPT与富电子有机磷酸盐相结合,构建了一系列宽范围和快速响应的有机−无机杂化光致变色晶态材料,在一定程度上优化了电子给体−受体杂化体系的光致变色性能[28-29]。最近,该课题组又将TPT引入到镧系复合物体系中,首次发现了镧系材料在室温下可逆的光致变色和光磁响应性能,实现了室温光生自由基精确调控单分子磁性行为,从而丰富了电子给体−受体杂化体系功能[30]。然而,通过有效调变电子给体−受体的界面关系,优化电子给体−受体的内部电子行为,实现分子间CT和ET的微妙平衡,进而实现有机−无机电子给体−受体杂化体系光致变色性能的可控性,目前还处于初级阶段。
基于上述考虑,我们将具有优异缺电子特性和多齿螯合配位能力的2,4,6-三(2-吡啶基)-1,3,5-三嗪(2-TPT)电子受体与 2,5-呋喃二羧酸(2,5-H2FCA)电子给体,借助不同配位模式的金属进行杂化,得到了3个2-TPT基配合物:[Zn(2-TPT)(2,5-FCA)](1)、[Cd(2-TPT)(2,5-FCA)]·1.5H2O(2)和[Mn(2-TPT)(2,5-FCA)](3)。考察了金属差异对于电子给体−受体间界面关系的调变,进而实现了对杂化体系内部分子间电子行为和光致变色性能的有效调控。
1 实验部分
1.1 试剂与仪器
2-TPT购自上海麦克林生化科技有限公司。2,5-H2FCA和Mn(NO3)2·4H2O购自阿拉丁试剂公司。Zn(NO3)2购自成都华夏化学试剂有限公司。Cd(NO3)2·4H2O购自国药集团化学试剂有限公司。N,N-二甲基甲酰胺(DMF)购自天津市科密欧化学试剂有限公司。所有试剂和溶剂均未经进一步纯化。
所用仪器有UitimaⅣ-I85型粉末X射线衍射仪(PXRD,Mo Kα,λ=0.071 073 nm,40 kV,40 mA,2θ=5°~40°)、Varian660-IR型傅里叶变换红外分光光度计、Rigaku UItimaⅣ-185型紫外可见光谱仪、Bruker A300-10/12型电子顺磁共振谱仪(EPR)、Kratos AXIS ULTRA型X射线光电子能谱仪(XPS,Al Kα,λ=0.835 7 nm)、Perkin Elmer型元素分析仪、HCT-2型差热热重分析仪、紫外光源(300 W,365 nm)。
1.2 配合物的制备
将 2-TPT(0.015 g,0.05 mmol)、2,5-H2FCA(0.011 g,0.07 mmol)、Zn(NO3)2(0.010 g,0.05 mmol)和 DMF(3 mL)置于50 mL烧杯中,在室温下混合,搅拌30 min使其充分混合。将混合溶液倒入10 mL的反应釜中,密封,在110℃加热48 h。待反应釜冷却至室温后,经抽滤、洗涤和干燥,得到配合物1,为黄色片状晶体,产率为58%(基于2-TPT)。配合物1(C24H14ZnN6O5)的元素分析计算值(%):C 54.16,H 2.63,N 15.80;实验值(%):C 54.67,H 2.57,N 15.71。红外光谱(KBr压片,cm−1):1 633(m),1 576(m),1 559(m),1 534(s),1 483(w),1 378(m),1 336(s),1 256(w),1 216(w),1 184(w),1 155(w),1 091(w),1 041(w),1 022(w),1 009(w),961(w),853(w),809(w),796(w),769(m),680(w),663(w),628(w),580(w)。将Zn(NO3)2分别替换为 Cd(NO3)2·4H2O(0.015 g,0.05 mmol)和 Mn(NO3)2·4H2O(0.013 g,0.05 mmol),其余步骤不变,即可合成配合物2和3。配合物2为浅黄色片状晶体,产率为66%(基于 2-TPT)。2(C48H34Cd2N12O13)的元素分析计算值(%):C 47.58,H 2.83,N 13.87。实验值(%):C 47.82,H 2.69,N 13.65。红外光谱(KBr压片,cm−1):1 611(m),1 592(m),1 574(m),1 551(s),1 534(s),1 473(w),1 439(w),1 381(s),1 364(s),1 257(w),1 213(w),1 156(w),1 096(w),1 047(w),1 025(w),1 006(w),951(w),853(w),813(w),771(m),666(w),626(w),572(w),524(w)。配合物3为橘色片状晶体,产率为60%(基于2-TPT)。3(C24H14MnN6O5)的元素分析计算值(%):C 55.29,H 2.71,N 16.12。实验值(%):C 55.13,H 2.62,N 16.19。红外光谱(KBr压片,cm−1):1 634(m),1 596(m),1 569(m),1 554(s),1 529(s),1 486(w),1 475(w),1 390(m),1 376(m),1 355(m),1 325(s),1 290(w),1 251(w),1 214(w),1 191(w),1 151(w),1 093(w),1 042(w),1 008(w),962(w),852(w),779(m),677(w),618(w),582(w),503(w)。
1.3 配合物晶体结构的测定
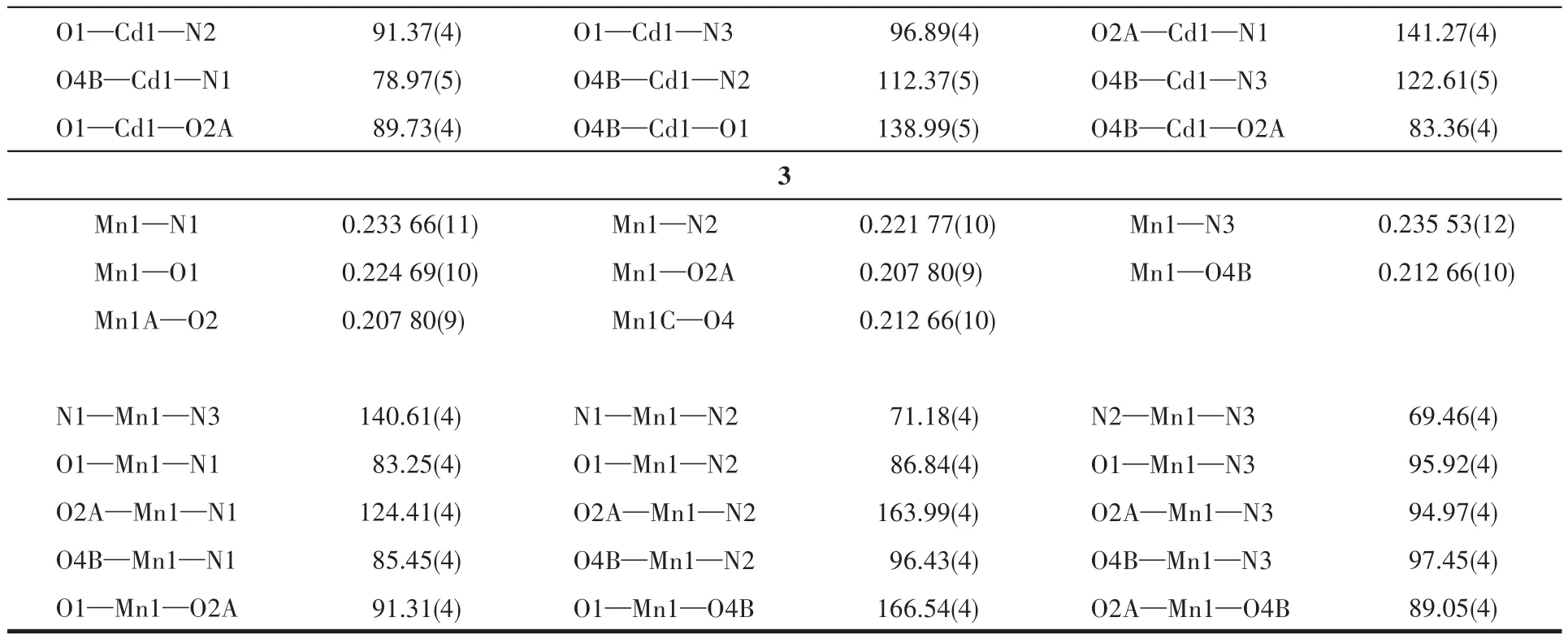
配合物1~3的晶体结构测定利用Oxford Gemini型单晶X射线衍射仪进行,采用经石墨单色器单色化的Mo Kα射线(λ=0.071 073nm)作辐射源,以ω-φ扫描方式收集衍射点。非氢原子用直接法解出,并对其坐标及各向异性热参数用全矩阵最小二乘法修正。氢原子的位置由理论加氢或寻找傅里叶峰得到,并使用固定的各向同性热参数加入结构精修。所有计算使用SHELXTL程序包和Olex2进行。所有非氢原子均进行了各向异性精修。晶体数据和结构精修参数列于表1,部分键长和键角列于表2,氢键数据列于表3。
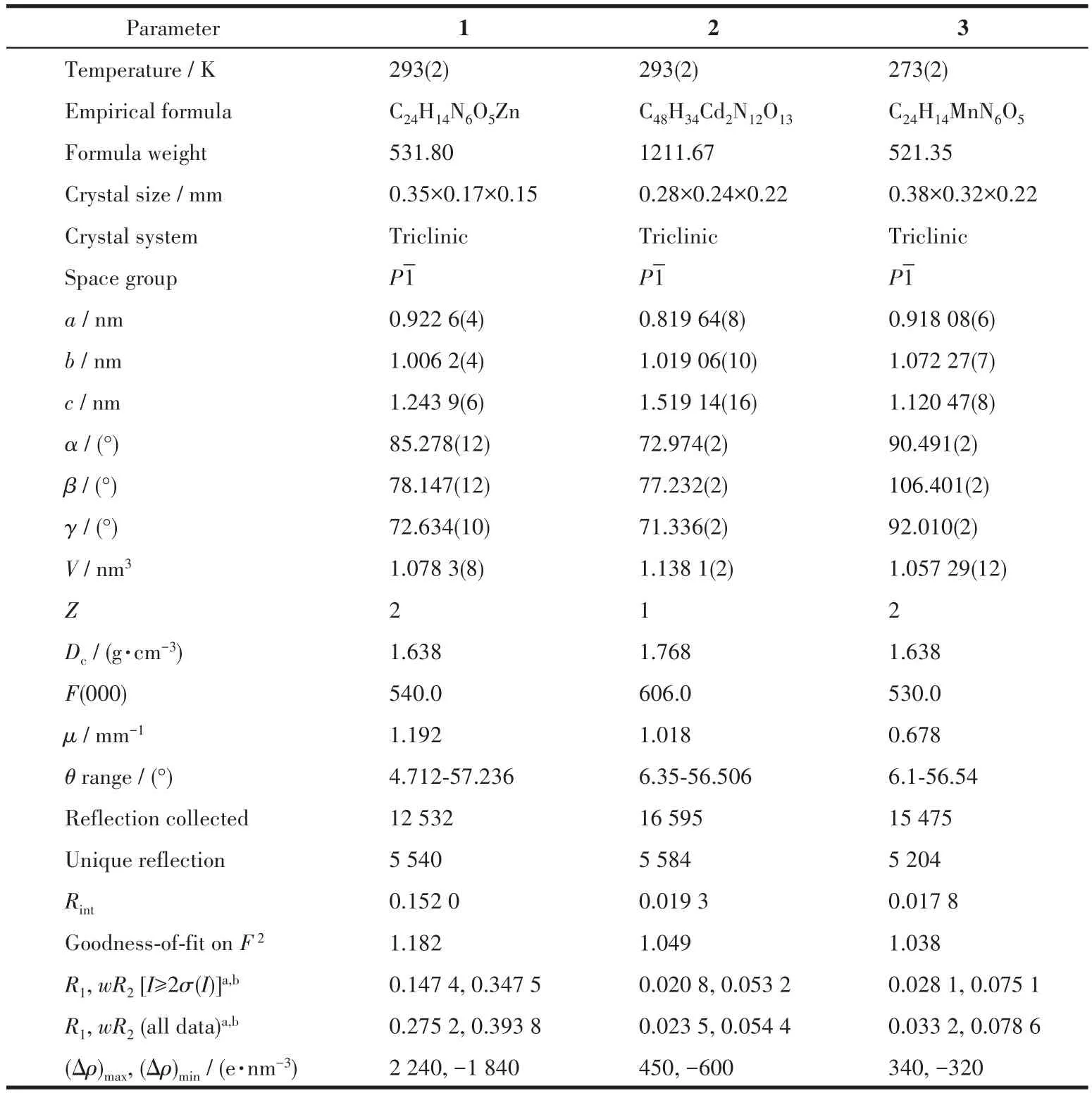
表1 配合物1~3的晶体学数据和精修参数Table 1 Crystallographic data and refinement parameters of complexes 1⁃3
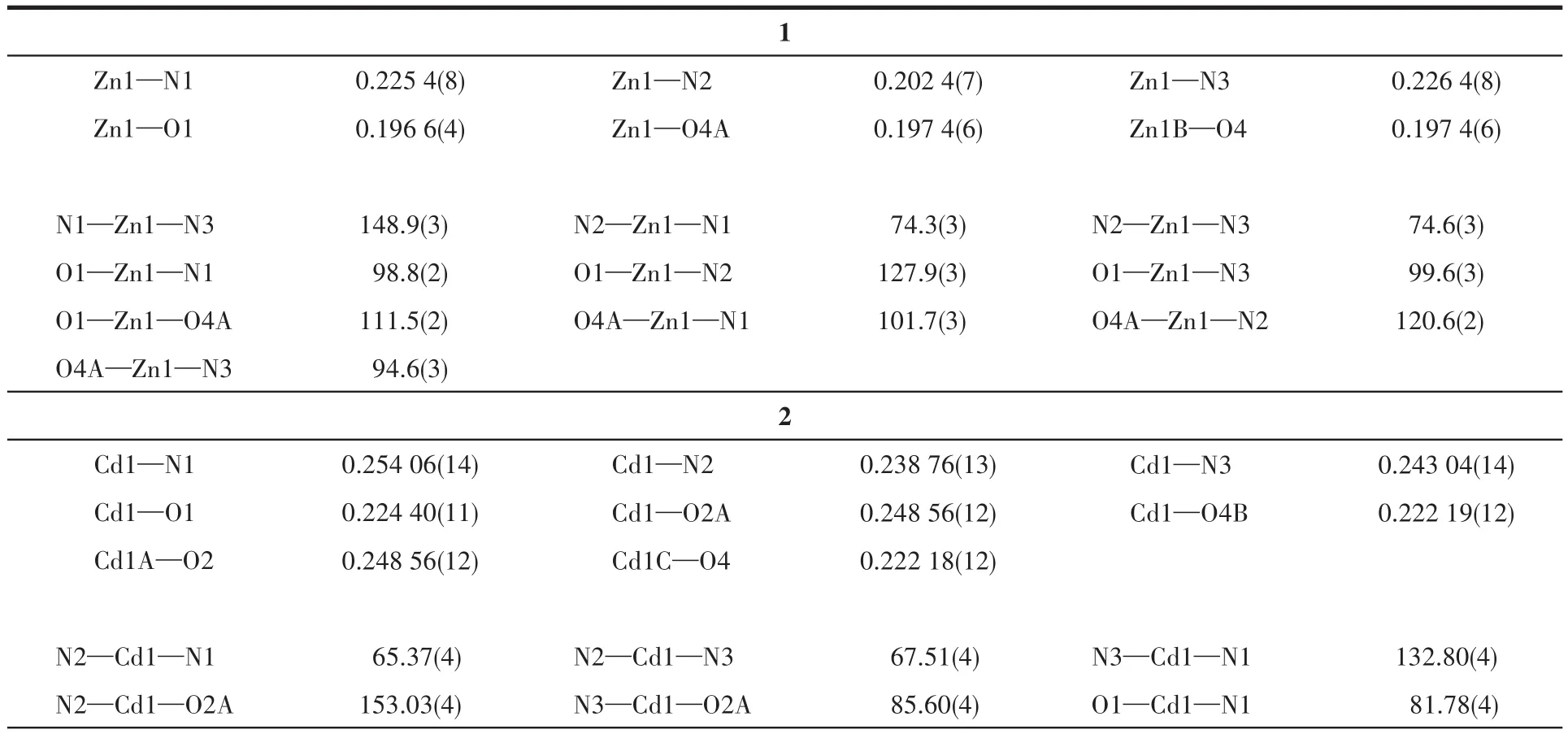
表2 配合物1~3的部分键长(nm)和键角(°)Table 2 Selected bond lengths(nm)and angles(°)for complexes 1⁃3
续表2
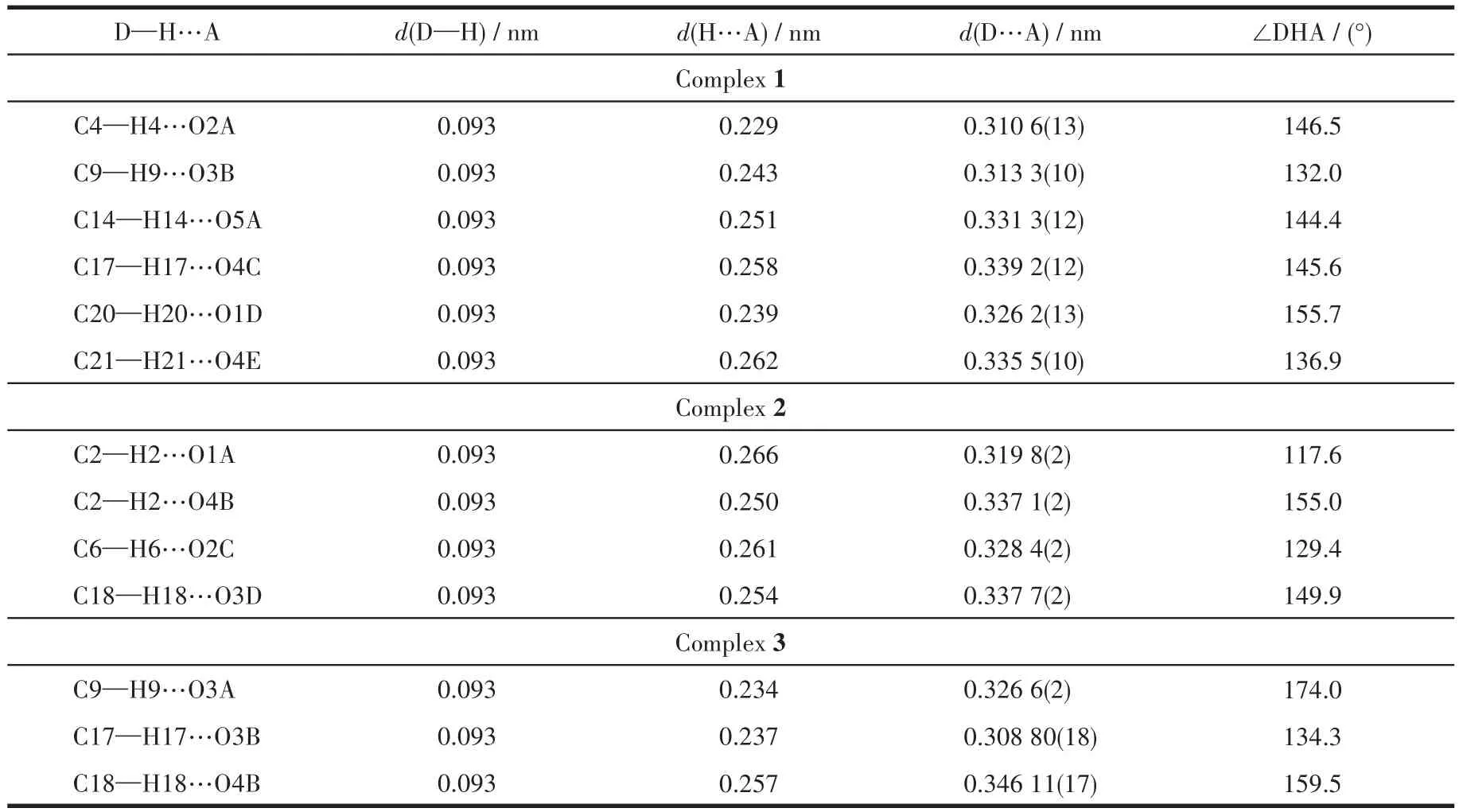
表3 配合物1~3的氢键参数Table 3 Hydrogen bond parameters for complexes 1⁃3
CCDC:2096882,1;2096883,2;2096884,3。
2 结果与讨论
2.1 配合物1~3的晶体结构
配合物1属于三斜晶系P1空间群,不对称结构单元包含1个晶体学独立的Zn(Ⅱ)离子、1个中性2-TPT分子和1个2,5-呋喃二羧酸阴离子(2,5-FCA2−)(图1a)。中心金属Zn(Ⅱ)离子是五配位的严重扭曲三角双锥构型,赤道平面上的3个配位点被来自1个2-TPT的3个N原子(N1、N2、N3)占据,轴向方向的2个配位点被来自2,5-FCA2−的羧基氧原子(O2、O4A)占据。Zn—O 和 Zn—N 键长分别在0.196 6(4)~0.197 4(6)nm和0.202 4(7)~0.226 4(8)nm之间,N—Zn—N和 O—Zn—N键角分别处于74.3(3)°~148.9(3)°和 94.6(3)°~127.9(3)°范围内,O—Zn—O键角为111.5(2)°。所有键长和键角都在正常范围内,并且与文献报道的2-TPT基锌配合物相一致[31]。2,5-FCA2−的O原子均以单齿配位的模式通过金属Zn与2-TPT桥联形成一条无限的1D链状结构(图1b),2-TPT基本垂直于羧酸链。如图1c所示,2条链之间通过分子间π-π弱相互作用形成了2D超分子层状结构,这2条链错位平行,2-TPT一正一反分别与2条链垂直,链以一对一对的形式存在。同一条链前后之间又通过分子间π-π弱相互作用最终形成了一个3D超分子网络结构(图1d和1e)。
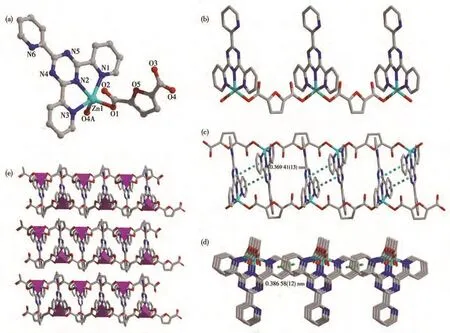
图1 配合物1的晶体结构:(a)不对称结构单元;(b)1D链状结构;(c)2D超分子层状结构;(d)同一条链前后存在π-π弱相互作用;(e)3D超分子网络结构Fig.1 Crystal structure of complex 1:(a)asymmetric unit;(b)1D chain structure;(c)2D supramolecular layer structure;(d)π-π weak interaction in front of and behind the same chain;(e)3D supramolecular network structure
配合物2属于三斜晶系P1空间群,不对称结构单元包含1个晶体学独立的Cd(Ⅱ)离子、1个中性的2-TPT分子、1个2,5-FCA2−阴离子和1.5个结晶水分子(图2a)。中心金属Cd(Ⅱ)是六配位的扭曲八面体构型,赤道平面上的4个配位点被来自2-TPT的3个氮原子(N1、N2、N3)和 1 个 2,5-FCA2−的羧基氧原子(O2A)占据,轴向方向的2个配位点被来自2,5-FCA2−的羧基氧原子(O2、O4B)占据。Cd—O 的键长在0.22218(12)~0.24856(12)nm之间,Cd—N的键长为0.238 76(13)~0.254 06(14)nm,N—Cd—N、O—Cd—N 和 O—Cd—O 键 角分 别 处 于 65.37(4)°~132.80(4)°、78.97(5)°~153.03(4)°和 83.36(4)°~138.99(5)°范围内。所有键长和键角都在正常范围内,并且与文献报道的2-TPT基镉配合物是一致的[32]。2,5-FCA2−的羧基O通过单齿和双齿2种不同的配位模式形成了一条1D的双排链(图2b)。在双排链中羧酸链和2-TPT基本呈垂直关系。双排链之间通过分子间π-π弱相互作用形成了2D的超分子层状结构(图2c和2d)。

图2 配合物2的晶体结构:(a)不对称结构单元;(b)1D双排链状结构;(c)2D超分子层状结构;(d)堆积图Fig.2 Crystal structure of complex 2:(a)asymmetric unit;(b)1D double-chain structure;(c)2D supramolecular layer structure;(d)packing diagram
配合物3同样属于三斜晶系P1空间群,不对称结构单元包含1个晶体学独立的Mn(Ⅱ)离子、1个中性的 2-TPT 分子和1个2,5-FCA2−阴离子(图3a)。配合物3与2的配位环境和连接模式完全一样,不再进行描述。但是由于中心金属的差异,3中双排链的扭曲程度较大,导致双排链间不存在弱相互作用(图3b和3c)。其中,Mn—O的键长处于0.207 80(9)~0.224 69(10)nm范围,Mn—N的键长在0.221 77(10)~0.235 53(12)nm 范围内,N—Mn—N、O—Mn—N和 O—Mn—O 键角分别在 69.46(4)°~140.61(4)°、83.25(4)°~163.99(4)°和 89.05(4)°~166.54(4)°范围内。所有键长和键角都在正常范围内,并且与文献报道的2-TPT基锰配合物是一致的[32]。
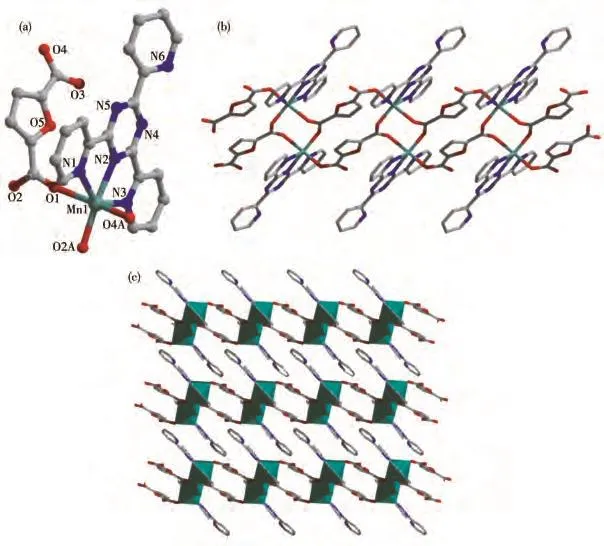
图3 配合物3的晶体结构:(a)不对称结构单元;(b)1D双排链状结构;(c)堆积图Fig.3 Crystal structure of complex 3:(a)asymmetric unit;(b)1D double-chain structure;(c)packing diagram
2.2 热重分析
配合物的热稳定性在实际应用中发挥着至关重要的作用,因此对1~3的热稳定性进行了探究。在氮气气氛的保护下(氮气流量为 100 cm3·min−1,升温速率为10 ℃·min−1,温度范围为30~800 ℃),通过热重−差示扫描量热法(TG-DSC)测定了配合物1~3的热稳定性。如图4所示,配合物1和3具有较高的热稳定性,TG-DSC曲线显示出二者只有一步的失重过程。1和3能够分别在361和368℃之前稳定存在,进一步加热导致骨架的坍塌,主要归因于有机配体的分解。配合物2的TG-DSC曲线显示出2个阶段的失重。第一个阶段的失重出现在94~208℃温度范围内,在差热曲线中也有明显吸热峰(171℃),对应于失去1.5个晶格水分子(实验值:3.69%,理论值:4.46%)。随后的重量损失大约从322℃开始,主要归因于有机配体分解导致的骨架坍塌。
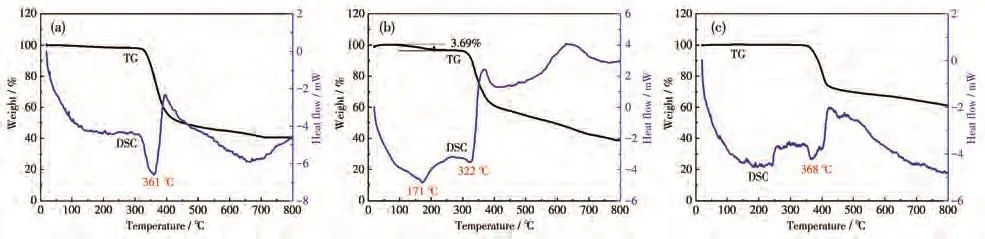
图4 配合物1(a)、2(b)和3(c)的TG-DSC曲线图Fig.4 TG-DSC curves of complexes 1(a),2(b),and 3(c)
2.3 粉末X射线衍射表征
如图5所示,配合物1~3的PXRD的实验图均与相应的理论模拟图基本吻合,表明合成的晶体1~3不含杂质,均为纯相。经汞灯(300 W,365 nm)辐照后的样品1P~3P的PXRD图与原样品1~3的PXRD相比,出峰位置和强度基本一致,表明光照前后晶体结构均没有发生变化,说明1~3的变色行为不是由结构变化引起的。
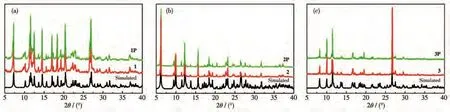
图5 配合物1(a)、2(b)和3(c)的PXRD图Fig.5 PXRD patterns for complexes 1(a),2(b),and 3(c)
2.4 红外光谱表征
通过对配合物1~3的红外分析(图6)可知:在3 500 cm−1附近较宽的强吸收峰是实验过程中H2O分子中H—O的伸缩振动引起的;在3 000~3 300 cm−1范围内出现的弱吸收峰,是由缺电子受体2-TPT的吡啶环和2,5-FCA2−阴离子的呋喃环上的C—H键伸缩振动导致的;在1 611~1 634 cm−1范围内出现的中等强度的吸收峰是由分子内C=O双键的伸缩振动引起的;在1 325~1 596 cm−1范围内出现的强吸收峰归因于2-TPT分子中C=N和C=C双键的伸缩振动;在951~1 390 cm−1范围内出现的弱吸收峰是2-TPT分子中吡啶环的C—C、C—N键弯曲振动引起的;900 cm−1以下的吸收峰是2-TPT分子平面外的C—H弯曲振动。对比配合物1~3光照前后的红外光谱可以看出,其出峰位置和强度均未发生明显变化,说明在光照前后3个配合物的官能团都没有发生变化。

图6 配合物1(a)、2(b)和3(c)的红外光谱图Fig.6 IR spectra of complexes 1(a),2(b),and 3(c)
2.5 变色行为的紫外可见吸收光谱分析
如图7所示,在汞灯(300 W,365 nm)辐照下,配合物1~3表现出截然不同的光致变色行为。配合物1和2表现出肉眼可见的颜色变化,分别从黄色和浅黄色变为黑色(1P)和银灰色(2P),二者都是从1 s开始变色,变色饱和时间分别为8和10 min。光照变色饱和样品1P和2P能够长时间稳定存在,在黑暗空气环境中分别放置约15和10 d,基本可以恢复起始颜色,表明二者的变色和褪色过程是可逆的。为了研究1和2的光致变色行为,对样品进行了时间依赖的紫外可见(UV-Vis)吸收测试。如图7a和7b所示,1在200~500 nm和2在200~450 nm范围内吸收带归因于芳香有机组分2-TPT的n-π*和π-π*跃迁。1和2经过光照后在可见光区内出现了新的特征吸收峰:1P的特征吸收峰在522、566、619和805 nm,2P的特征吸收峰出现在573、628和800 nm。随着光照时间的延长,样品颜色逐渐加深,时间依赖的UV-Vis吸收光谱中显示的吸收强度逐渐增加,当样品颜色不再变化时,UV-Vis吸收光谱的吸收强度也趋于饱和。这些特征吸收峰与文献报道的TPT自由基吸收类似,因此配合物1和2的光致变色性能可能归因于光诱导的分子间ET并伴随(2-TPT)−•阴离子自由基的生成。相比1和2优异的光致变色行为,3的起始颜色为橘色,对应于其在可见光区的吸收带380~650 nm。经光照1 h后,配合物3P的紫外吸收有一定程度增强,但其颜色在光照前后基本没有变化,表明其不具有光致变色性能。

图7 配合物1(a)、2(b)和3(c)的光致变色图(插图)和时间依赖的UV-Vis吸收光谱图Fig.7 Photographs showing photochromic behaviors(Inset)and time-dependent UV-Vis absorption spectra of complexes 1(a),2(b),and 3(c)
2.6 EPR表征
根据上述对比分析光照前后配合物1和2的PXRD图和红外谱图,可以排除光解或结构变化引起的光致变色行为。配合物1和2在光照后在UVVis吸收光谱图中出现了新的特征吸收峰,我们推测是由于光诱导ET产生有色自由基。为了进一步确认配合物1和2的自由基变色机理,对样品进行了EPR表征。如图8所示,光照前的样品1和2没有出现明显的自由基信号,光照后的样品1P和2P分别在g=2.000 9和2.001 6处出现了强的自由基信号,进一步证明了1和2是由光照过程中产生了有色自由基而引起的变色。将变色后的样品1P和2P经过长时间的黑暗处理后,强的自由基信号基本消失。此外,1P的自由基信号明显强于2P,与变色后颜色的深浅和UV-Vis吸收峰的强度相一致。然而,配合物3在光照前后都有较宽的信号,这可能归因于金属Mn自身的磁性信号,而不是有色自由基的信号。根据前期我们对分子间CT和ET及二者关系的研究[33-36],配合物3不变色的原因主要在于较强的分子间CT对光诱导分子间ET的抑制作用。

图8 配合物1(a)、2(b)和3(c)的EPR图Fig.8 EPR spectra of complexes 1(a),2(b),and 3(c)
2.7 XPS表征
为了进一步揭示变色机理,并明确分子间ET通道,对1~3光照前后的样品进行了XPS测试。如图9和10所示,光照前后1和2中C1s结合能均未发生明显变化,1中Zn2p和2中Cd3d结合能也不存在明显差异;值得注意的是,O1s总体向高的结合能方向移动(1:从530.41 eV移动到530.88 eV;2:从530.98 eV移动到531.78 eV),N1s总体向低的结合能方向移动(1:从399.33 eV移动到398.90 eV;2:从405.28 eV移动到404.70 eV)。这些结果证明了光照前后配合物1和2中N原子得到了电子,O原子失去了电子,进一步说明发生了从富电子给体2,5-FCA2−阴离子到缺电子受体2-TPT的分子间ET。如图11所示,光照前后3中C1s、N1s、O1s和Mn2p结合能均未发生明显变化,进一步证明光照后未发生光诱导分子间ET。

图9 配合物1光照前后的XPS谱图Fig.9 XPS spectra of complex 1 before and after light irradiation

图10 配合物2光照前后的XPS谱图Fig.10 XPS spectra of complex 2 before and after light irradiation
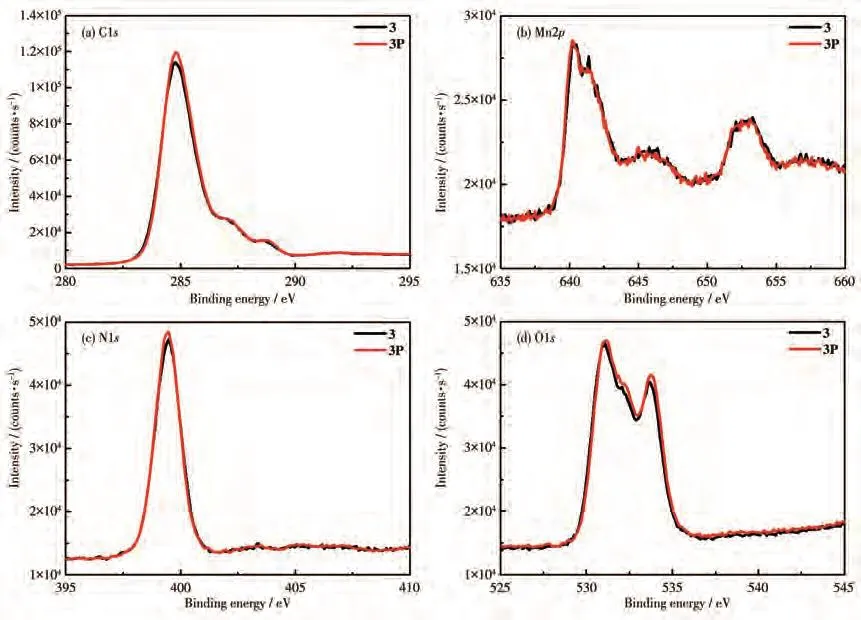
图11 配合物3光照前后的XPS谱图Fig.11 XPS spectra of complex 3 before and after light irradiation
2.8 界面关系分析
为了深入分析配合物1~3的光致变色性能的差异,对三者中电子给体和受体间的界面关系进行了深入分析和对比。如图12所示,配合物1~3的羧酸基团上的O原子到2-TPT中三嗪环上N原子的最近距离分别为 0.291 07(9)、0.334 31(3)和 0.349 53(2)nm,这与3个配合物的光响应性能呈现对应关系。其中,1的光致变色性能(较快的光响应速率和明显的颜色对比度)显著优于2,主要归因于1中的O—N距离比2中的短。3不具有光致变色行为,主要归因于较长的O—N距离。因此,3个配合物光响应行为的差异主要归因于不同配位模式金属所引起的晶体堆积模式和电子给体−受体间界面关系的不同,体现了金属可以有效调控分子间的电子行为和光致变色性能。此外,光致变色后的样品1P和2P能够稳定存在,除了(2-TPT)−·阴离子自由基自身的稳定性外,还与2个配合物中存在较强的π-π相互作用(质心距离:0.369 41(13)、0.386 58(12)nm,1;0.371 84(4)nm,2)有关。
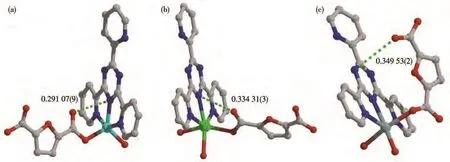
图12 配合物1(a)、2(b)和3(c)的电子给体和受体间的界面接触Fig.12 Interfacial contacts between electron donor and acceptor in complexes 1(a),2(b)and 3(c)
3 结论
将缺电子受体2-TPT、富电子给体2,5-H2FCA分别与不同类型的金属离子(Zn、Cd和Mn)配位,制备了3个2-TPT基配合物。通过单晶X射线衍射、PXRD、FT-IR、UV-Vis、TG-DSC、EPR和XPS等手段对配合物1~3的结构和光致变色性能进行了表征,并详细地探究了其光致变色机理。配合物1~3表现出明显差异的光诱导分子间电子转移和光致变色性能,主要体现在光响应速率和颜色对比度2个方面。研究表明,3个配合物明显差异的光致变色行为,主要归因于不同类型的金属及其配位模式的差异所引起的不同晶体堆积模式和电子给体−受体间界面关系,体现出了金属对于光致变色性能的调控作用。
猜你喜欢
无机化学学报(2022年9期)2022-09-16
井冈山大学学报(自然科学版)(2022年2期)2022-03-31
原子与分子物理学报(2022年3期)2022-03-05
辽宁科技大学学报(2021年2期)2021-07-22
青岛大学学报(工程技术版)(2019年2期)2019-09-10
人工晶体学报(2019年5期)2019-06-18
当代陕西(2019年6期)2019-04-17
分析化学(2018年2期)2018-03-02
读写算·教研版(2016年8期)2016-05-07
中学化学(2015年8期)2015-12-29
